

A picture is worth a thousand endpoints: high-content imaging in drug discovery
Posted: 12 September 2016 | Debra Taylor (MRC Technology), Emilie Bureau (MRC Technology), Janet Brownlees (MRC Technology) | No comments yet
Over the past few years several factors in the drug discovery industry have come together to drive an increased interest and subsequent surge in the use of high-content imaging. These include the high costs of attrition in the clinic due to lack of efficacy; increased hurdles and rising costs to get new drugs to market; a swing of the pendulum back to phenotypic-based screening; as well as a timely improvement in high-content imaging technology and analysis software…
High-content imaging is a very powerful technique that can link compound effects to physiological and morphological cellular changes in a robust and quantitative manner. However, while the hardware has been available for 10-15 years, it has taken some time for this technology to reach anywhere near its potential.
In the early days, assays had very simple readouts, such as cell number count or morphological change, and mainly used recombinant cell lines for secondary assays in screening cascades. Nowadays, assay development involves more complex cellular systems driven by a desire to monitor an increased number of endpoints in both fixed and live cells and also to make use of these high-content assays in primary screening campaigns. Results that are more translatable to the clinic can be yielded from more physiologically relevant human cells including primary cells or those derived from induced pluripotent stem cells.
However, having now achieved what could be regarded as real ‘high-content’ imaging, a new bottleneck has arisen: that of data storage and analysis, along with the handling and reporting of multiparametric data. Solutions for this are now being developed, which may actually be within reach for use by the common biologist, rather than specialist computer scientists. Importantly, the quantity of data generated from an assay can be tailored to address and answer scientific questions. It is apparent that high-content imaging is now having an impact on current drug discovery projects, and although it has yet to be proven in the clinic, given the power of this technology this may not be too distant a dream.
Drug Target Review has just announced the launch of its NEW and EXCLUSIVE report examining the evolution of AI and informatics in drug discovery and development.
In this 63 page in-depth report, experts and researchers explore the key benefits of AI and informatics processes, reveal where the challenges lie for the implementation of AI and how they see the use of these technologies streamlining workflows in the future.
Also featured are exclusive interviews with leading scientists from AstraZeneca, Auransa, PolarisQB and Chalmers University of Technology.
From target-based to phenotypic approaches
The pharmaceutical industry continually strives for ways to improve the drug discovery process in order to increase success rates in the clinic and to address those aspects that are known to be responsible for poor translational rates, such as efficacy and toxicity. From the 1980s to the late-2000s the industry focused on reductionist target-based drug discovery following the huge increase of genomic information from whole genome mapping. This has failed to live up to the expectations of increasing the numbers of successful drugs on the market1,2. The drug discovery industry appears to have come full circle and is once again showing increased interest in what could be termed classical phenotypic drug discovery, whereby viewing the system as a whole (be it cell or organism) there is an increased chance of generating more physiologically-relevant data than that gained from studying a single target.
In the past 15 years, however, most new chemical entities approved by the United States Food and Drug Administration have still come from target-based approaches with only a small percentage resulting from phenotypic screening2. In the future, however, it is likely there will remain a need for both approaches.
The increased interest in phenotypic screening and the desire to deliver more in-depth information from physiologically-relevant cells or tissues has furthered the need to develop better technology. Cell biologists have historically monitored cellular response by classical microscopy and, from standard microscopic examination of samples on slides, engineering advances have produced microscopes capable of imaging cells on microtiter plates in automatic mode. This has enabled researchers to record high numbers of images on a cell-per-cell basis and for the era of high-content imaging to evolve. With these advances in engineering technology over the past ten years or so came the need for more advanced analysis software, in order to extract maximal information from various cellular readouts and from small numbers of cells. It is now possible to access several hundred endpoints, but whether we actually use this, or even need to use this amount of information to answer every question is debatable.
How many endpoints?
A recent review from Singh et al showed that although there has been a steady increase in the number of papers published reporting the use of high-content imaging since 2000, the information content is much lower than its potential, with 83% of such papers reporting the use of only one or two endpoints3. Why is this the case? Is it because researchers have reduced the data or endpoints to be analysed, based on their own (probably biased) knowledge of the mechanism, to the minimal amount required to prove the hypothesis, or to identify the most active compound as they simply cannot handle the analysis of the large volume of data generated? For example, during a lead optimisation campaign, chemists want to have the key data presented in a succinct manner and data reports generally have to fit with corporate databases, where the recording of multiple endpoints may not fit with the normal method of data processing. Also, most groups do not have the correct infrastructure in place to deal with the vast amounts of data that the modern-high content imagers are capable of generating and so biological responses, which could be important in developing a more efficacious drug, may be being missed.
Is a two-endpoint assay sufficient for structure activity relationships?
At MRC Technology (MRCT) we have successfully used a two-endpoint imaging assay to drive chemistry structure activity relationships (SARs) for an Alzheimer’s project, the aim of which was to reduce the phosphorylation of the microtubule binding protein, tau. The project was to develop potent and specific inhibitors of the kinase microtubule affinity regulating kinase (MARK) which phosphorylates an epitope within the microtubule binding domain of tau, Ser262, a key site in the regulation of tau binding to microtubules. In this assay both primary rat cortical and human neurons derived from hippocampal stem cells (PhoenixSongs Biologicals, Inc., United States) were used and the images were taken on the InCell 2000 (GE Healthcare). The first level software, Workstation, was sufficient to write an efficient analysis script to generate the two endpoints of interest – phosphorylated tau using intensity-of-fluorescence staining by a specific antibody and nuclear cell count as a measure of toxicity (Figure 1). As only two endpoints were used, data transfer from the imager and subsequent pharmacological analysis was easily performed using the standard software packages, Microsoft Excel and GraphPad Prism.
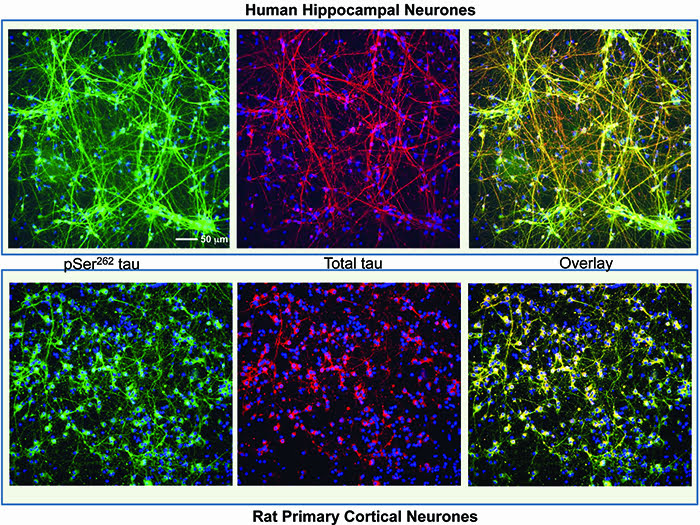
Figure 1: Human hippocampal neuronal cells (top panel) and rat primary cortical cells (bottom panel) stained with a tau antibody specific for phosphorylation on Ser262 (Life Technologies) shown in green, total tau antibody (DAKO) shown in red and nuclei stained with Hoescht 34580 shown in blue. The overlay image shows co-localisation of the phospho and total tau signal. The staining for phosphorylated tau is mainly in the cell body in the rat neurones but more evenly distributed across the whole neurone in the human cells. Images were taken on an InCell 2000 High Content Imager (GE Healthcare) using a 20x objective.
Incorporating additional endpoints
MRCT and academics collaborate closely on projects and assays, which are often developed taking observations made by the academic into consideration. In some cases an assay may already be available, but it requires miniaturisation and optimisation to enable it to be used in drug screening. Academic labs’ imaging work tends to be carried out using standard microscopes meaning data can be limited, however this is now changing with more academic labs purchasing high-content imagers. If it is potentially amenable to higher throughput, MRCT will convert a slide-based assay into a plate-based, higher throughput and higher content assay. As an example, a project investigated osteoporosis therapeutics where the assay was designed to measure a decrease in osteoclastogenesis. The ‘gold standard’ method of monitoring osteoclastogenesis is to measure tartrate-resistant acid phosphatase (TRAP) enzymatic activity by standard light microscopy5. MRCT developed a high-content imaging assay using RAW264.7 cells to monitor differentiation of macrophages into osteoclasts. Endpoints included the size and number of cells and also classifications based on numbers of nuclei per cell, which increase as the cells become larger and more differentiated into osteoclasts.
Although this assay represents the key phenotypic aspects of differentiation, the assay was modified to incorporate a fluorescent readout of TRAP levels. Therefore, one assay monitors both phenotypic changes and enzymatic activity (Figure 2). The images were analysed using a bespoke algorithm written using the more sophisticated software, GE Developer. Data handling for this osteoclast assay was more involved with the increase in the endpoint numbers and also the classification of cells.
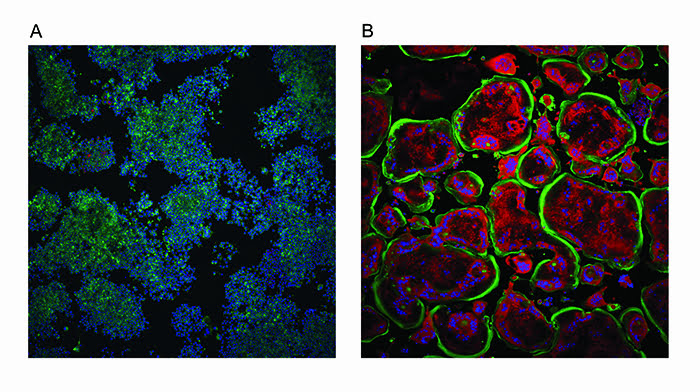
Figure 2: Osteoclastogenesis assay. Undifferentiated mouse macrophage RAW264.7 cells (A) were treated with nuclear factor κB ligand (RANKL) to promote differentiation into osteoclasts (B). Cells were stained with ActinGreen 488™ ReadyProbes (Thermo Scientific) to show the peripheral actin ring in green and Hoechst 34580 to show nuclei in blue. The red fluorescence represents TRAP activity which increases with differentiation as does the number of nuclei per cell and the size of the cell (substrate was naphtol As-Mx phosphate disodium salt). Images were taken on an InCell 2000 High Content Imager (GE Healthcare) using a 10x objective.
Phenotypic screening – many endpoints, a cellular picture
Early reviews on high-content imaging described it as a technology rarely applied to screening campaigns, but used more for target validation and secondary screening6. However, there has been a move within the pharmaceutical industry over the past few years to incorporate phenotypic screening into drug discovery programmes. Because of this MRCT and others are now building the IT infrastructure and robotics (including hotel incubators and robotic arms) to enable phenotypic cellular screening using high-content imagers. An example of such an assay, which we are carrying out for the Alzheimer’s Research UK Dementia Consortium, is screening for compounds that clear cellular aggregates of a protein, TAR DNA-binding protein (TDP-43). Aggregates of this protein are found in many neurodegenerative diseases and mutations have also been identified in amyotrophic lateral sclerosis. The aggregates are thought to be detrimental – or at least indicative that there are other negative cellular effects contributing to progression of the disease.
A phenotypic screen of aggregate clearance in a fluorescently-tagged engineered TDP-43 cell line7 has identified compounds that are target- and pathway-agnostic, but produced the desired phenotype. An example of the typical cellular imaging produced in this screen is shown in Figure 3. This high content assay measures many cellular parameters including aggregate levels (average and total per cell), cell number, nuclear morphology and measures of cellular and mitochondrial toxicity.
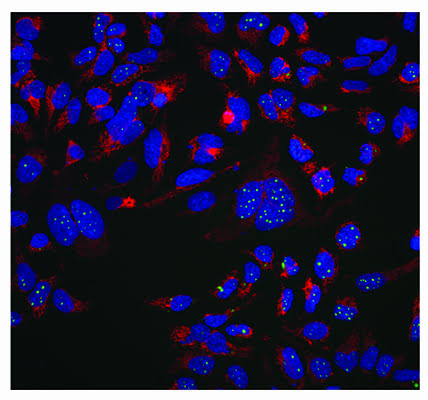
Figure 3: Aggregate clearance assay for neurodegenerative disease indication. A protein which aggregates in neurodegenerative disease (TDP-43) is labelled with a GFP tag and stably expressed in an inducible system in Hek293 cells. MRCT compound libraries have been screened to identify compounds which reduce this aggregation in a phenotypic imaging screen. The green channel shows aggregated GFP labelled protein, the red is a mitochondrial potential dye (TMRM) which stains healthy mitochondria and the blue is nuclei stained with Hoechst 34580. Images were taken on an InCell 2000 High Content Imager (GE Healthcare) using a 20x objective.
Although it is not essential to identify targets for FDA approval and chemists are more open to performing SAR using phenotypic screens, identification of the target and/or mechanistic pathway is always desirable for follow up drug discovery efforts. Target identification from phenotypic screens is incredibly difficult and approaches including screening of annotated compound sets underpinned by strong bioinformatics analysis are being used in attempts to achieve this. Another approach is labelling sufficiently potent compounds to enable interacting proteins to be pulled down from cell lysates, followed by subsequent analysis and possible identification by mass spectrometry.
Cellular toxicity
In all cell assays it is important to monitor toxicity, especially for inhibition assays, since any decrease in readout could be due solely to cell death and completely independent of the mechanism being studied. A particularly useful feature of high-content imaging is the ability to include toxicity readouts with the phenotypic readout to enable faster and more informed chemical triaging of hits. In addition to the simultaneous toxicity readout – and in order to build more detailed annotations and enable early decision making around screen hits – various MRCT compound sets have been screened in a high-content imaging toxicity assay. This assay has four endpoint readouts (cell number, membrane potential, mitochondrial potential and intracellular calcium levels) and is based on the assay developed by O’Brien et al,4. Toxicity information is used in conjunction with phenotypic screen endpoints and results from screening annotated compound sets covering nodes in many key biological pathways to try and build more in depth knowledge around the biology of the assay readout.
Data analysis
As high-content imaging evolves and assays become even more complicated and generate even more data, the bottleneck in this process at MRCT and many other institutions is the data transfer, storage, handling and interpretation of meaningful results from a plethora of endpoints. To enable analysis and presentation of more complex data, commercial software designed for handling and analysing large data sets was investigated. For this we collaborated with Genedata (Switzerland) and evaluated their Screener software – in particular, their High Content Screening (HCS) module. This software provides both image and data management solutions and allows the review of images directly aligned to the calculated results, providing instant confirmation of phenotypic changes and biological events. With the venture into phenotypic screening, this software package has become incredibly useful and is now embedded within our high-content imaging processes. There is a desire within MRCT to use as much information from assays as we need in order to make an informed decision; be this two, several or many endpoints. Initial analysis of the full data sets would not be possible without this data handling software, be it commercial or, as is the case for other institutions, in-house generated.
Physiologically relevant cell types
With the development of induced pluripotent stem cell (iPSC) technology over the past few years there is, of course, a desire within the screening community to try and use these more physiologically relevant cell types. However, the use of such cells is still in the very early stages and although some groups have tried to do this in a high-throughput manner, there are still issues restricting the use of primary stem cells. These issues are mainly that of cost; supply (and actually being able to get enough cells to screen); reproducibility; and robust characterisation of the differentiated cell type in the case of stem cells. For the time being there would appear to be, despite much effort and desire, an air of quiet acceptance to screen in less specialised cells but conduct hit confirmation and follow up using iPSCs or primary cells. In addition, although high-content imaging reduces the number of cells required, there is still an issue of growing cells in smaller microtiter plates, which is not always straightforward or indeed possible.
By harnessing the power of high-content imaging, which can generate the numbers of endpoints required to paint a cellular picture, and using the new generation of more user-friendly analysis and data handling tools, we anticipate a continued rise in the use of this technology to solve more complex problems. Hopefully, rewards will be reaped from the efforts in developing this technology and it will not be too long before a compound or biologic identified from high-content imaging will reach the market and thus confirm the importance of this technology in the drug discovery process.
Biography
 JANET BROWNLEES is a Team Leader within the Compound Profiling and Cellular Pharmacology Group at the Medical Research Council Technology (MRCT) Centre for Therapeutics Discovery (CTD) located at the Stevenage Bioscience Catalyst science park just outside London. She gained a biochemistry degree and PhD at the Queen’s University of Belfast followed by post-docs in neurodegeneration at the Institute of Psychiatry, King’s College London and also University College London. Janet worked in various departments at GlaxoSmithKline in Harlow from 2003 responsible for teams developing cellular assays for target validation, screening and hit validation in early-stage drug discovery before joining MRCT in 2010.
JANET BROWNLEES is a Team Leader within the Compound Profiling and Cellular Pharmacology Group at the Medical Research Council Technology (MRCT) Centre for Therapeutics Discovery (CTD) located at the Stevenage Bioscience Catalyst science park just outside London. She gained a biochemistry degree and PhD at the Queen’s University of Belfast followed by post-docs in neurodegeneration at the Institute of Psychiatry, King’s College London and also University College London. Janet worked in various departments at GlaxoSmithKline in Harlow from 2003 responsible for teams developing cellular assays for target validation, screening and hit validation in early-stage drug discovery before joining MRCT in 2010.
References
- Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011; Jul; 10 (7): 507-19
- Eder J, Sedrani R, Wiesmann C. The discovery of first-in-class drugs: origins and evolution. Nat Rev Drug Discov. 2014; Aug; 13 (8): 577-87
- Singh S, Carpenter AE, Genovesio A. Increasing the Content of High-Content Screening: An Overview. J Biomol Screen. 2014; Jun; 19 (5): 640-50
- O’Brien PJ, Irwin W, Diaz D, Howard-Cofield E, Krejsa CM, Slaughter MR, et al. High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch Toxicol. 2006; Sep; 80 (9): 580-604
- Burstone MS. Histochemical demonstration of acid phosphatase activity in osteoclasts. J Histochem Cytochem. 1959; Jan; 7 (1): 39-41
- Bickle M. High-content screening: a new primary screening tool? IDrugs 2008; Nov; 11 (11): 822-6
- Budini M, Romano V, Quadri Z, Buratti E, Baralle FE. TDP-43 loss of cellular function through aggregation requires additional structural determinants beyond its C-terminal Q/N prion-like domain. Hum Mol Genet. 2015; Jan 1; 24 (1): 9-20
Related topics
Analytical techniques, Assays, Cell-based assays, Drug Discovery, High-content assays, Screening, toxicology