

Using genetics to identify drug targets for fibrotic lung diseases
Posted: 4 June 2020 | Richard Allen (University of Leicester) | No comments yet
Researchers recently conducted a genome-wide association study (GWAS) to identify factors that increase susceptibility to the incurable lung condition idiopathic pulmonary fibrosis (IPF). Here, Dr Richard Allen shares some of the study’s key findings.

Idiopathic pulmonary fibrosis (IPF) is a deadly lung disease that currently has no cure. In individuals with IPF, we believe the lungs experience repetitive microinjury (for example from inhaled dusts, viral infections, etc,) and an abnormal repair response leading to the deposit of scar tissue.1 This scar tissue builds up, resulting in stiffening of the lungs and impairing of gas transfer between the lungs and bloodstream. Over time, this leads to reduced lung health and a poorer quality of life, as half of patients diagnosed with IPF die within three to five years.1,4
There are two drugs (pirfenidone and nintedanib) licenced for the treatment of IPF, but these only slow disease progression and most patients experience side effects when taking these drugs.2,3 There are around 6,000 new cases of IPF in the UK and around 30,000-40,000 new cases in the US each year. The US National Institute of Health reports the worldwide prevalence of IPF to be 13-20 per 100,000 people,5 suggesting there are 1-1.5 million cases of IPF worldwide.
Identifying more risk signals will help to refine our understanding of which biological pathways are important for IPF risk and therefore guide the development of new treatments”
IPF is the most common of the interstitial lung diseases (ILD), a heterogeneous group of more than 150 conditions characterised by inflammation and scarring of the interstitial regions of the lungs.6 In March of this year, following the results of a recent trial,7 the US Food and Drug Administration (FDA) approved the use of nintedanib to treat patients with other non-IPF chronic fibrosing ILD.8 This includes individuals with a progressive phenotype of hypersensitivity pneumonitis, autoimmune ILD and idiopathic nonspecific interstitial pneumonia. Therefore, understanding the condition is not only required when investigating possible IPF treatments, but also potentially identifying treatments for a range of fibrotic lung diseases.
Drug Target Review has just announced the launch of its NEW and EXCLUSIVE report examining the evolution of AI and informatics in drug discovery and development.
In this 63 page in-depth report, experts and researchers explore the key benefits of AI and informatics processes, reveal where the challenges lie for the implementation of AI and how they see the use of these technologies streamlining workflows in the future.
Also featured are exclusive interviews with leading scientists from AstraZeneca, Auransa, PolarisQB and Chalmers University of Technology.
The reason people develop IPF is still unknown; however, by utilising genetics, we can reveal particular biological pathways and specific genes and proteins that contribute to disease risk. This is important for treatment discovery for two reasons: 1) by identifying the pathways that are driving disease risk we may be able to develop treatments that target these pathways (and targets supported by genetic evidence are twice as likely to be successful during drug development9) and 2) we may be able to find subsets of patients who are more likely to respond to certain treatments (ie, personalised medicine).
A GWAS is a hypothesis-free approach where genetic variants from across the genome are tested for their association with a trait of interest (Figure 1). When genetic variants associated with disease risk are identified, we can then investigate the functional effects of the variant to discover important biological processes in disease risk and potential drug targets. Large sample sizes are required to detect these genetic effects, which are usually of modest magnitude. Modest genetic effect sizes, however, can translate to large clinical effects if the genes that they implicate encode druggable targets. Despite this advantage, recruiting the large sample sizes necessary can prove difficult for rarer diseases, such as for IPF.
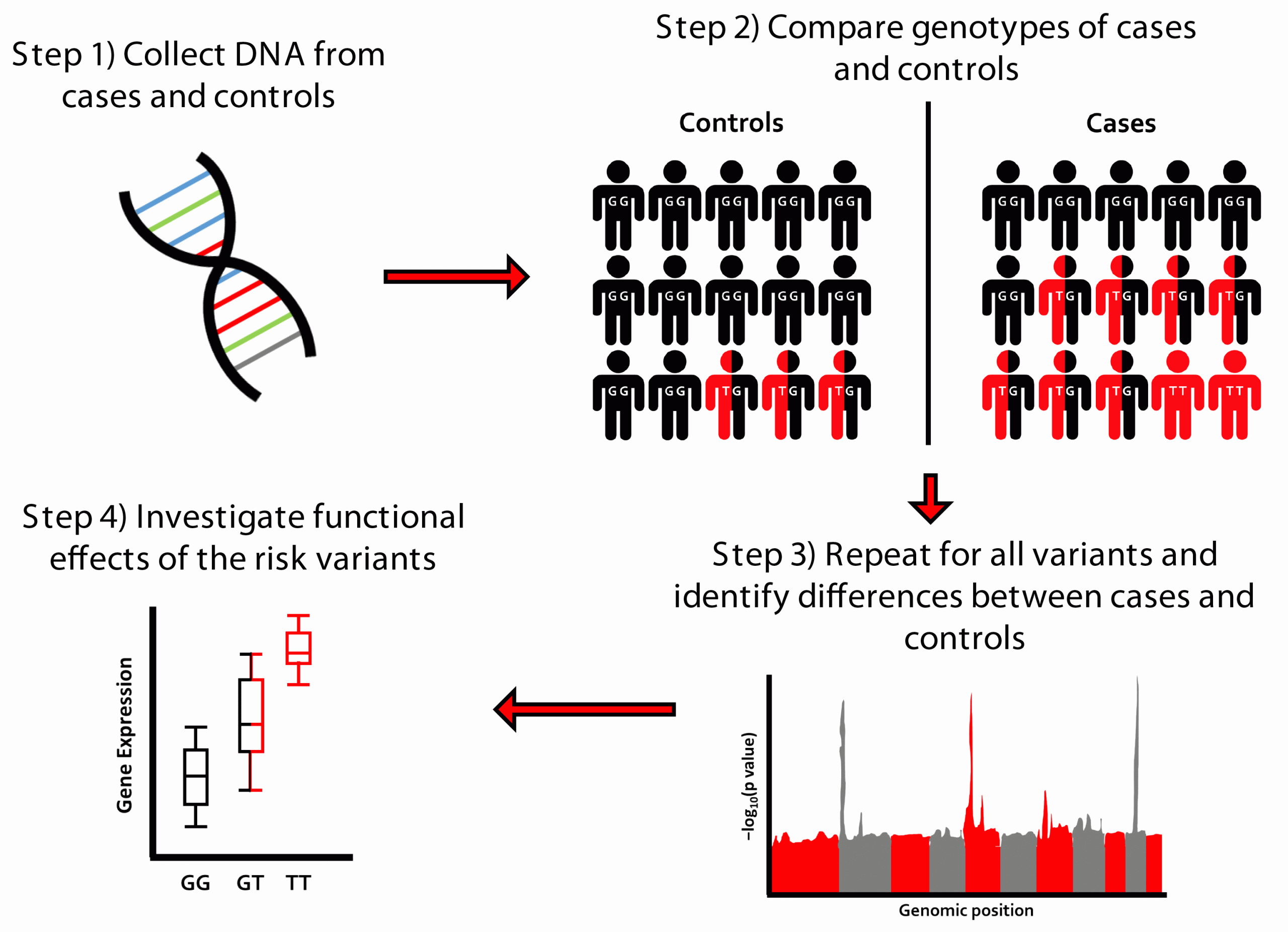
Figure 1: Overview of genome-wide association study (GWAS) design.
The largest known risk factor of IPF is a genetic variant called rs35705950. In the general European population, around 79 percent of people have a GG genotype at this variant, around 20 percent have GT and one percent have TT. For each copy of the T allele at this variant, the risk of developing IPF increases fivefold.10 This variant lies in the promoter region of a gene called MUC5B, which encodes a component of mucus called mucin 5B. Individuals with the T allele show higher levels of expression of MUC5B and it is thought that the increased mucus production swamps the cilia in the airways, impairing clearance of pathogens that in turn trigger the fibrotic response.
Three previously published IPF risk GWAS on European samples identified a number of biological processes important in disease risk.11-13 These include host defence (such as the MUC5B variant previously described), signalling (specifically in genes that regulate TGFβ signalling), cell-cell adhesion (such as desmoplakin which plays a role in structural integrity of the epithelium) and telomere maintenance (as individuals with IPF tend to have shorter telomeres than age-matched controls).
One novel IPF risk signal was associated with increased expression of the DEPTOR gene in lung tissue”
In our recent study, we combined all of the cases and controls from these three previous GWAS into one larger international GWAS.14 We identified new regions of the genome associated with IPF risk and tested whether these were genuine associations in two independent case-control studies. In total, we were able to compare the DNA of over 4,000 individuals with IPF to the DNA of 20,000 individuals without IPF. This presented three new regions of the genome that are associated with IPF risk and which contain the DEPTOR, MAD1L1 and KIF15 genes.
One novel IPF risk signal was associated with increased expression of the DEPTOR gene in lung tissue. The importance of mTORC1 signalling via 4E-BP1 to regulate collagen deposition by mediating TGFβ signalling in the lungs was recently reported.15 DEPTOR inhibits mTOR signalling, suggesting that higher levels of DEPTOR leads to decreased mTOR signalling which results in the deposition of more collagen and build-up of scarring in the lungs. mTOR inhibitors are licenced for cancer therapy16 and clinical trials of mTOR inhibitors for treatment of IPF are taking place (NCT01725139, NCT03502902, NCT01462006). The other two novel IPF risk signals lie in spindle assembly genes. While KIF15 and MAD1L1 have not been implicated in IPF pathogenesis previously, these genes may contribute to telomere maintenance.17
In total, there are 14 regions of the genome known to be associated with IPF. This enables us to begin narrowing down which particular genes, proteins and biological pathways drive IPF susceptibility. Identifying more risk signals will help to refine our understanding of which biological pathways are important for IPF risk and therefore guide the development of new treatments. Through a risk score approach (ie, by combining the effects of multiple genetic variants) it is possible to show that there are potentially hundreds of common genetic variants associated with IPF that we cannot yet detect with our current sample sizes.
There are limitations to these kinds of studies. Firstly, DNA sequencing studies have shown that rare variants (particularly in telomere18 and surfactant19 genes) are significant in IPF risk. However, rare variants are not well-captured using the genotype-array data used in our study. Secondly, most of the IPF GWAS undertaken to date have been restricted to individuals of European genetic ancestry to reduce confounding effects. This bias towards European samples currently exists across genetic studies for all complex diseases and we know relatively little about the genetic determinants of IPF risk in non-Europeans. This may lead to increased healthcare inequalities between ancestry groups for medical approaches that build on genetics, such as precision/stratified medicine, risk prediction and pharmacogenetics.20
This study also highlights the power of collaboration. IPF is not a common condition, meaning it can be difficult to recruit the number of individuals needed for a GWAS. This study was only possible due to the input of collaborators from over 30 institutions (including academia, clinical and industry) from six countries. Hopefully, through continued and expanded collaborations, we will be able to maximise the potential of genetic studies to further unravel IPF pathobiology and identify new and much-needed drug targets.
Author biography
Dr Richard Allen is an Action for Pulmonary Fibrosis Mike Bray research fellow at the University of Leicester. His research focuses on understanding the genetic basis of idiopathic pulmonary fibrosis risk and identifying disease subtypes through large-scale statistical analyses. He graduated from the University of York with a Mathematics and Statistics BSc and completed his Medical Statistics MSc and PhD in genetic epidemiology at the University of Leicester.
References
- Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378(19):1811-1823.
- National institute for health and care excellence (2013) pirfenidone for treating idiopathic pulmponary fibrosis. NICE technology appraisal guidance (TA282). availiable at: https://www.nice.org.uk/guidance/TA282.
- National institute for health and care excellence (2016) nintedanib for treating idiopathic pulmonary fibrosis. NICE technology appraisal guidance (TA379). available at: https://www.nice.org.uk/guidance/ta379.
- Ley B, Collard HR, King Jr TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine. 2011;183(4):431-440.
- National Institute of Health. Genetics home reference: Idiopathic pulmonary fibrosis. https://ghr.nlm.nih.gov/condition/idiopathic-pulmonary-fibrosis#. Accessed 11th March, 2020.
- Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine. 2006;174(7):810-816.
- Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718-1727.
- FDA News Release. FDA approves first treatment for group of progressive interstitial lung diseases. https://www.fda.gov/news-events/... Accessed 9th March, 2020.
- Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47(8):856-860.
- Zhu QQ, Zhang XL, Zhang SM, et al. Association between the MUC5B promoter polymorphism rs35705950 and idiopathic pulmonary fibrosis: A meta-analysis and trial sequential analysis in caucasian and asian populations. Medicine (Baltimore). 2015;94(43):e1901.
- Noth I, Zhang Y, Ma S, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. The Lancet Respiratory Medicine. 2013;1(4):309-317.
- Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45(6):613-620.
- Allen RJ, Porte J, Braybrooke R, et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of european ancestry: A genome-wide association study. The Lancet Respiratory Medicine. 2017.
- Allen RJ, Guillen-Guio B, Oldham JM, et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine. 2020;201(5):564-574.
- Woodcock HV, Eley JD, Guillotin D, et al. The mTORC1/4E-BP1 axis represents a critical signaling node during fibrogenesis. Nature communications. 2019;10(1):6.
- Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. Journal of hematology & oncology. 2019;12(1):71.
- Coe BP, Lee EH, Chi B, et al. Gain of a region on 7p22. 3, containing MAD1L1, is the most frequent event in small‐cell lung cancer cell lines. Genes, Chromosomes and Cancer. 2006;45(1):11-19.
- Dressen A, Abbas AR, Cabanski C, et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: A candidate gene sequencing study. The Lancet Respiratory Medicine. 2018.
- Petrovski S, Todd JL, Durheim MT, et al. An exome sequencing study to assess the role of rare genetic variation in pulmonary fibrosis. American journal of respiratory and critical care medicine. 2017;196(1):82-93.
- Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51(4):584.
Related topics
Disease research, DNA, Drug Targets, Genetic analysis, Genomics
Related conditions
Idiopathic pulmonary fibrosis (IPF), Pulmonary fibrosis
Related organisations
US Food and Drug Administration (FDA)