

In vitro platforms for de-risking nephrotoxicity during drug development
Posted: 25 March 2020 | Colin Brown (Newcells Biotech), Sandy Primrose (Newcells Biotech) | 1 comment
If not correctly assessed, drug candidates with undesirable safety issues may progress through clinical development, resulting in costly failures later in the development process. Given that many drugs fail in clinical trials due to nephrotoxicity, this article describes the importance of understanding kidney transporter function in drug development, how current cell line and animal models are insufficient and how suitable pre-clinical testing with human tissue models can reduce this risk.
THE KIDNEY is a significant organ for the clearance of drugs from the body: 32 percent of the top 200 prescribed drugs undergo renal elimination. Clearance involves two physiological processes: glomerular filtration and tubular modification of the filtrate by tubular secretion and reabsorption. Glomerular filtration is a passive process in which drugs or metabolites that are not bound to proteins present in blood plasma (free fraction) diffuse from the blood into the primary urine. Tubular secretion and reabsorption are active processes mediated by solute carriers and ATP-binding cassette transporters, primarily in the proximal tubular cells of the renal cortex.
The main influx transporters such as organic anion transporters 1 and 3 (OAT1 and OAT3) and organic cation transporter 2 (OCT2) are located in the basolateral membrane, whereas the major efflux transporters such as multidrug and toxin extrusion (MATE1), multidrug associated resistance protein (MRP) and P-glycoprotein (P-gp) are located in the apical membrane of the proximal tubules (Figure 1).
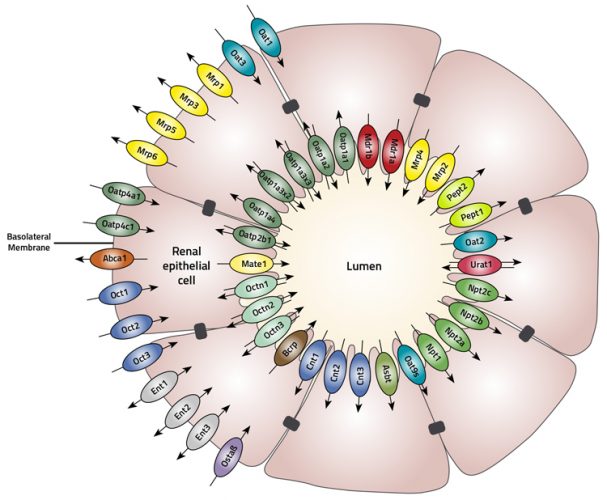
Figure 1: The proximal tubule is comprised of a cylinder of polarised epithelial cells joined together with an apical located tight junctional complex. Proximal tubule cells express a large number of transport proteins that are asymmetrically located either to the apical or basolateral membrane.
While renal transporters generally enhance renal elimination, certain transporters such as OAT4, PEPT2 and SGLT2 and the receptor-mediated endocytic complex megalin-cubulin located on the apical side of the proximal tubule cells facilitate reabsorption of some drugs. The combined action of apical and basolateral transporters determines the rate at which drugs move from the blood into the urine or from urine to blood.1
Drug Target Review has just announced the launch of its NEW and EXCLUSIVE report examining the evolution of AI and informatics in drug discovery and development.
In this 63 page in-depth report, experts and researchers explore the key benefits of AI and informatics processes, reveal where the challenges lie for the implementation of AI and how they see the use of these technologies streamlining workflows in the future.
Also featured are exclusive interviews with leading scientists from AstraZeneca, Auransa, PolarisQB and Chalmers University of Technology.
Tubular secretion and nephrotoxicity
Because tubular secretion is essentially a transporter mediated process, anything that interferes with it can affect the half-life of a drug (if the interaction occurs at the basolateral uptake transporter) or can cause nephrotoxicity (if the interaction occurs at the apical transporter). Drug-drug interactions are widespread and are often a direct result of interference with tubular secretion. For example, cimetidine and pyrimethamine reduce the renal clearance of metformin. Cimetidine inhibits OCT2 – and hence the uptake of metformin by proximal tubule cells – whereas pyrimethamine inhibits MATE1 or MATE1/2-K and hence efflux from the cells.2 In this example, cimetidine and pyrimethamine would be contraindicated in diabetic patients taking metformin.
Nephrotoxicity can occur when there is an imbalance between influx and efflux of a drug, and the pyrimethamine-induced accumulation of metformin is a good example of this process. Nephrotoxicity can also occur in the absence of drug-drug interactions. The anticancer drug cisplatin causes renal toxicity due to its high rate of accumulation in the tubular epithelial cells mediated by OCT2 and rate-limiting efflux via transporters MATE1 and MATE2-K.3 These issues mean that during drug development, the ability to predict whether a drug is likely to be cleared by kidney secretion and, if so, determine if it will accumulate in the kidney epithelial cells or if other drugs will inhibit secretion is crucial. The US Food and Drug Administration (FDA) has recognised the importance of understanding these issues and in 2010 issued guidance in the form of a whitepaper, which required drug companies to understand the mechanisms of drug transport and potential drug-drug interactions if the renal clearance of their candidate molecule was greater than 25 percent of total clearance.
The FDA guidance has led to an almost 100 percent uptake by industry of screening candidate molecules using cell lines transfected with single renal transporters. This approach has been enormously successful in understanding which transporters may be necessary in determining the renal clearance of a molecule. However, they are essentially ‘jigsaw pieces’ and do not provide information on the importance of individual transporters in drug handling by the intact proximal tubule, nor are they able to provide unambiguous information on potential drug-drug interactions. For this, an alternative approach that allows the study of drug transport and drug-drug interactions in an intact proximal tubule is required. Because access to fresh human kidney can be limiting, most studies have used cells that have been immortalised following infection with oncogenic viruses or hTERT; for example, HK-2, RPTEC and ciPTEC. However, cell lines of this type do not differentiate and lose transporter expression in culture (Table 1).
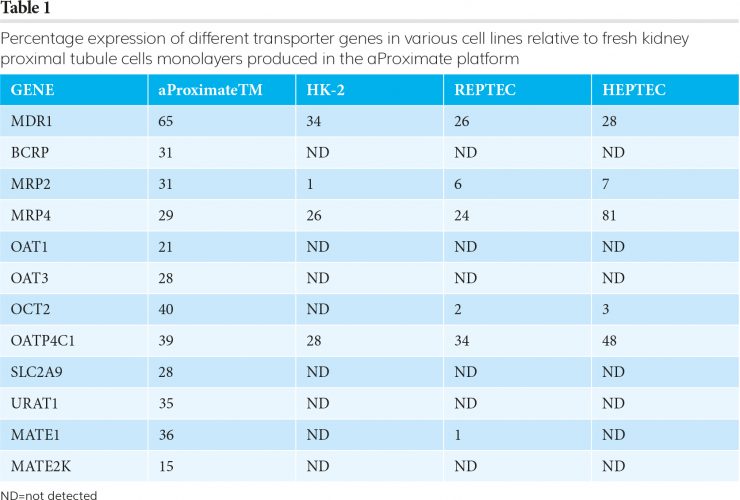
Because of the aforementioned inherent issues associated with permanent cell lines, a far better approach is to use primary tubule cells freshly isolated from ethically sourced transplant-grade kidney tissue.4 When these cells are grown as monolayers, they retain much of the function and differentiation Figure 1 of the original tissue. In particular, they express all the relevant transporters (aProximateTM in Table 1) and have an appropriate transepithelial electrical resistance (TEER). When the flux of mannitol and creatinine was measured across the monolayers, the values for creatinine were far larger than for mannitol. This is consistent with creatinine flux being mediated by transporters through the cells
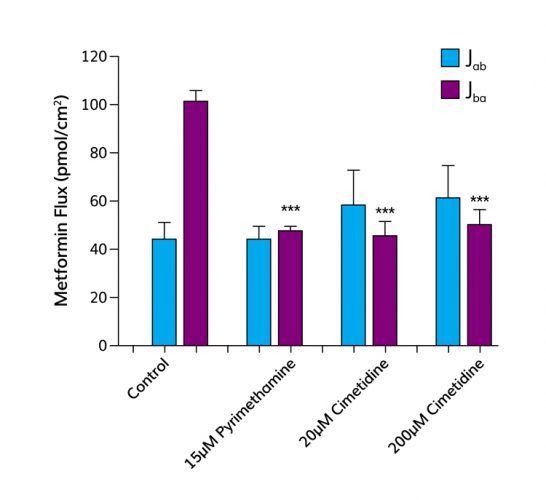
Figure 2: The absorptive (Jab) and secretory (Jba) fluxes of metformin (10цM) were measured across monolayers of human proximal tubule cells grown on TranswellTM filter supports. The secretory flux of metformin was significantly greater than the absorptive flux revealing net secretion from basolateral to apical compartment. In the presence of pyrimethamine (the substrate for MATEs) the secretory flux of metformin was significantly reduced, abolishing net secretion. Similarly, in the presence of cimetidine (the substrate for OCT2) on the basolateral side, the secretory flux of metformin was significantly reduced, abolishing net secretion of metformin.
whereas mannitol passes between the cells. Since the proximal tubule monolayer was developed, a wealth of validation data has been generated with over 50 new chemical entities (NCEs) supplied by pharmaceutical companies. The monolayers also exhibit functional expression of megalin and cubulin, the key uptake transporters of large molecules, and so can be used to study the transport of biologicals and oligonucleotides. Proximal tubule cells that form monolayers and remain differentiated for up to 10 days have also been generated from rats, as well as other pre-clinical species. This means that Table 1 an NCE can be introduced to the human and rat systems in parallel, to identify potential species differences in renal handling of a candidate before animal screening takes place.
A key test of the platform is its ability to identify potential drug-drug interactions. As previously mentioned, cimetidine interferes with the uptake of metformin by proximal tubule cells and pyrimethamine with efflux of metformin. In vivo, this leads to a significant increase in systemic exposure (AUC) and a decrease in renal clearance. Figure 2 shows that these interactions can be predicted in vitro, as in vivo metformin undergoes net secretion across the proximal tubule monolayer, but in the presence of pyrimethamine the net secretion of metformin is significantly inhibited. This is because metformin and pyrimethamine compete for efflux mediated by MATEs. This result in vitro translates in vivo to a decrease in renal clearance and an increase in AUC. Similar data can be obtained for interactions between metformin and cimetidine. In vivo co-administration of cimetidine and metformin results in an increase in AUC and a decrease in renal clearance. Figure 2 shows that the in vitro platform can predict this in vivo drug-drug interaction; the addition of cimetidine results in a significant decrease in the net secretion of metformin.
A long-established method of determining the pharmacokinetics and risk of toxicity of a potential new drug is to obtain data in animal models. The fact that only two percent of NCEs are abandoned at the pre-clinical stage due to nephrotoxicity but 18-27 percent during clinical studies in humans highlights the inadequacy of these animal models. The inadequacy originates, in part, from differences in the abundance of kidney transporters in different species. In a study of the distribution of 19 transporters across five species (human, monkey, dog, rat and mouse), 16 of the transporters occurred at higher levels in the pre-clinical test species.5 The differences ranged from 2.8-fold to 8.2-fold. There are also differences between the level of transporters in males and females of the same species, and therefore in drug toxicity.
Another problem is sequence differences in key transporters. For example, human OAT1 has serine at residue 203 whereas in all the standard animal models it is alanine. The human S203 variant has a much higher affinity for certain drugs than the animal A203 variant. The consequence of this variation led to the FDA refusing to approve adenofir for the treatment of HIV infections because of nephrotoxicity.6
Once a potential new drug has satisfactorily completed pre-clinical safety studies, it will usually be tested in a phase 1 clinical trial involving healthy human volunteers to determine the safe dose range and any potential side effects. For drugs that are metabolised or excreted by the kidney, the FDA has recommended that the levels of six biomarkers (Table 2) should be measured in the urine of triallists to aid the detection of tubular injury.
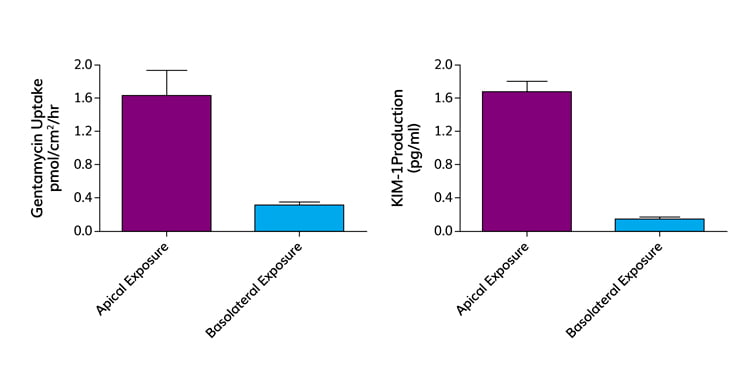
Figure 3: Gentamycin toxicity in vivo is mediated by megalin-cubulin mediated reabsorption of gentamycin from the primary urine. In human proximal tubule cell monolayers, gentamycin is preferentially absorbed into the proximal tubule cell across the apical membrane. Apical uptake of gentamycin is correlated with an increase in release of KIM-1, a biomarker of renal proximal tubule damage. In contrast, both the uptake and release of KIM-1 are almost zero if gentamycin challenge is from the basolateral side.
Gentamycin is a nephrotoxic antibiotic, primarily because of cytotoxic effects resulting from uptake by the megalin and tubulin complex on proximal tubule epithelial cells, which in turn elicits release of the KIM-1 biomarker into urine. This effect was replicated using our platform (Figure 3). In particular, compounds that are substrates for the megalin and cubulin transporters competitively reduce the uptake of gentamycin from the apical (urine) side of the monolayers. The reduction in uptake correlates with a reduction in the expression of the KIM-1 biomarker.
Recently, the potential of the aProximate system to accurately predict renal toxicity was evaluated with a panel of 36 chemically diverse drugs. Testing of the compounds was performed blind and Figure 4
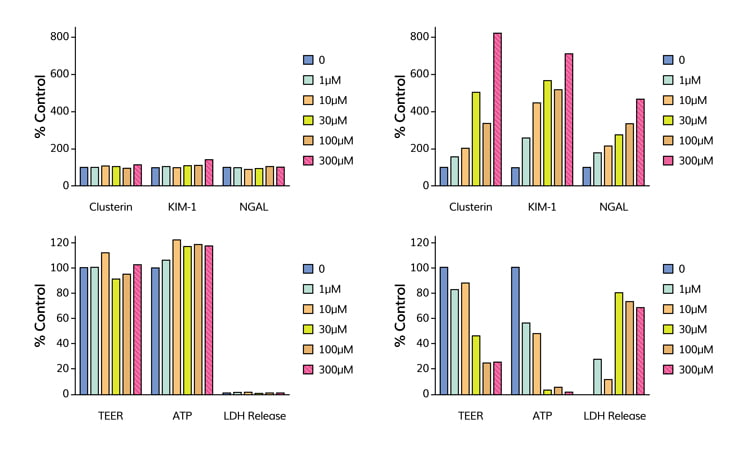
Figure 4: High-content screen of endpoints of toxicity. The endpoints included production of biomarkers: clusterin, KIM-1 and NGAL together with transepithelial resistance (TEER), ATP-content of cells and LDH release. Cell monolayers were challenged for 72 hours with a range of concentrations (0-300чM) of either the non-nephrotoxic acetylsalicylic acid (left panel) or nephrotoxic pemetrexed (right panel).
shows some of the data generated. In agreement with the published in vivo data, acetylsalicylic acid showed no toxic responses over the range of concentrations tested, whereas pemetrexed showed a dose-dependent release of kidney injury biomarkers and significant changes in measures of cell health. Subsequently, it was revealed that of the 36 compounds tested, 19 were known to be nephrotoxic. Applying ROC analysis to the dataset suggested that best predictivity (sensitivity, selectivity and overall accuracy) came if changes in the production of two biomarkers were used as the endpoint of toxicity. These data points suggest that the aProximate model is highly predictive of outcome in vivo and outperforms both animal studies and in vitro models in its accuracy.
Conclusions
The platform described above generates data on drug-drug interactions that is predictive of clinical outcome, maps onto FDA guidance on transporter data and biomarkers of kidney injury and outperforms animal studies.
About the authors
 Sandy Primrose has a BSc in Applied Microbiology from the University of Strathclyde and a PhD in Microbiology from the University of California (Davis). After 10 years in academia, he moved to industry where his career variously involved production of biopharmaceuticals, development of new assays for drug development and new diagnostics for various cancers. He is Chairman of Newcells Biotech.
Sandy Primrose has a BSc in Applied Microbiology from the University of Strathclyde and a PhD in Microbiology from the University of California (Davis). After 10 years in academia, he moved to industry where his career variously involved production of biopharmaceuticals, development of new assays for drug development and new diagnostics for various cancers. He is Chairman of Newcells Biotech.
 Colin Brown has a BSc and PhD in Physiology from the University of St Andrews. His research interests are the development of primary renal cell models to study species variation in ADMET and the use of human proximal tubule cells to study drug interactions with the kidney. He is Director of ADMET for Newcells Biotech.
Colin Brown has a BSc and PhD in Physiology from the University of St Andrews. His research interests are the development of primary renal cell models to study species variation in ADMET and the use of human proximal tubule cells to study drug interactions with the kidney. He is Director of ADMET for Newcells Biotech.
References
- Meyer zu Schwabedissen HE, Verstuyft C, Kroemer HK, Becquemont L, Kim RB. Human multidrug and toxin extrusion 1 (MATE1/SLC47A1) transporter functional characterization, interaction with OCT2 (SLC22A2) and single nucleotide polymorphisms. American Journal of Physiology – Renal Physiology 2010; 298: F997-1005.
- Feng B, LaPerle Jl, Chang G, Varma MV. Renal clearance in drug discovery and development: molecular descriptors, drug transporters and disease state. Expert Opinion in Drug Metabolism and Toxicology 2010; 6: 939-52.
- Yonezawa A, Inui K. Organic cation transporter OCT/SLC22A and H(+)/organic cation antiporter MATE/SLC47A are key molecules in nephrotoxicity of platinum agents. Biochemical Pharmacology 2011; 81, 563-8.
- Brown CDA, Sayer R, Windass AS, Haslam IS, De Broe ME, D’Haes PC, Verhulst A. Characterisation of human tubular cell monolayers as a model of proximal tubular xenobiotic handling. Toxicology and Applied Pharmacology 2008; 233, 428-38.
- Basit A, Radi Z, Vaidya VS, Karasu M, Prasad B. Kidney cortical transporter expression across species using quantitative proteomics. Drug Metabolism and Disposition 2019; doi: 10.1124/dmd.119.086579.
- Zou L, Stecula A, Gupta A, Prasad B, Chien H-C, Wah Yee S, Wang L, Unadkat JD, Stahl SH, Fenner KS, Giacomini KM. Molecular mechanisms for species differences in organic anion transporter 1, OAT1: implications for renal drug toxicity. Molecular Pharmacology 2018; 94, 689–99.
Related topics
Assays, Drug Development, In Vitro, In Vivo
Related organisations
Newcells Biotech, US Food and Drug Administration (FDA)
This article is very interesting! Congratulations to the Authors.