

Monoclonal antibody discovery: current status and future perspectives
Posted: 5 September 2018 | Dr. Stefan R. Schmidt | No comments yet
Since the first description of how to generate monoclonal antibodies was published in the 1970s, this field of research has grown tremendously. Currently, more than 70 full-length antibodies or fragments are approved by the major regulatory agencies globally. Looking back, a lot has changed since this historic discovery.
Initially the whole technology focused on utilising the immune system of small animals. Mice, rats or rabbits were injected with the respective antigen conjugated to a carrier protein like Keyhole Limpet Hemocyanin (KLH) and accompanied by an adjuvant. This complex antigen binds to membrane bound B-cell gamma globulins, crosslinking them, followed by internalisation and processing to reappear as a bound peptide in the class II Major Histocompatibility Complex (MHC II) on the B-cell surface. Interaction with the T-cell receptor on T-helper cells then releases cytokines that trigger the differentiation of the B-cell to secrete antibodies. Repeated injections over the course of a month boosts the antibody generation. Following that general principle, the antibody producing B-cells are then isolated from the spleen of the animals. In order to maintain antibody production for an extended period, the cells must be immortalised, which is achieved by fusing the B-cells with mouse myeloma cells that do not secrete antibodies of their own. The new cell resulting from that fusion process is called hybridoma and combines both the antibody specificity of the B-cell and the longevity and reproductivity of the myeloma cell. The myeloma cells are deficient of the hypoxanthine-guanine phosphoribosyltransferase (HGPRT) gene, which allows subsequent selection in a hypoxanthine-aminopterin-thymidine (HAT) medium, thus eliminating unfused myeloma cells. The fusion of these different cells is induced either by an electrical field or by chemical agents, such as polyethylene glycol (PEG). To obtain a monoclonal cell line, the pool of hybridomas must be diluted down to those individual cells that are true clones and thus precisely represent a single antibody type.
The basic concept of monoclonal antibodies is certainly quite elegant, but it suffers from some drawbacks. Firstly, the process is time consuming and requires live animals. Secondly, the method does not allow the screening and selection of antibodies by predefined conditions for binding. Thirdly, the resulting antibodies are rodent immunoglobulins, which are recognised by the human immune system but will only have limited therapeutic potential. Consequently, humanisation by transplanting the hypervariable loops of the rodent antibody into a human framework was developed to solve the issue.3 Finally, hybridoma cells are not ideally suited for production purposes as their expression level is rather low and they suffer from some genetic instability, due to the fact they are tetraploidic cells.
In vitro screening approaches
These limitations have led to variations of the initial technology being developed; most of which involve isolating the genes that define the variable domains of heavy and light chains of the antibody and generating highly complex libraries that are expressed recombinantly in different hosts. The most widely applied technology is called antibody phage display, which was first described in the early 1990s.4 Here, a coat protein of a filamentous phage incorporates a combinatorial library of rearranged heavy (VH) and light (VL) chains. In a panning process, binding phages are separated from nonbinding ones by washing and elution and amplified the by re-infection of bacteria. Standard molecular biology methods like sequencing the respective binding region and transferring the coding genes into an expression host, allow the production of antibodies for a variety of applications. The affinity can be further increased by mutagenesis of the isolated binders and subsequent repeated panning. Focusing the mutagenesis only on the complementarity-determining region (CDR) loops is preferable because it avoids disruptive mutations in the framework regions. As all six CDRs contribute to antigen binding, a stepwise process of mutagenesis can be advantageous. However, libraries of more than 1012 variants are difficult to integrate into standard phage displays.
An alternative ribosome display was developed that could avoid some of the disadvantages of phage display.5 Here, the starting point is the polymerase chain reaction (PCR) amplification of a cDNA library coding for single chain variable domains (scFv), introducing a T7 promotor, the ribosome binding site, and spacer sequences in place of a stop codon. After in vitro mRNA transcription and translation, the mRNA is not released from the ribosome due to the lack of a stop codon and the complex of mRNA, translated protein and ribosome stays connected as a complex. Complexes containing antigen binding scFvs are separated from non-binders in a classical panning procedure. Following chemical release of the complex components, the mRNA is isolated, reverse transcribed and again amplified by PCR to enter a second cycle. The whole process can be performed in vitro, independent of transformation, thus enabling faster and simpler methods, including the seamless integration of maturation by PCR.
In the context of display methods, surface display on microorganisms like yeast or bacteria should also be mentioned.6 The selection of binders of all surface display systems is no longer done on solid surfaces, but instead by fluorescent activated cell sorting (FACS), which simultaneously allows quantitative analysis of kinetic parameters. In the case of yeast display, Aga2p receptor of S. cerevisiae was chosen as a module to present scFv libraries on the cell surface. The passage through the secretion pathway enables typical post-translational processing like folding and is better suited than bacterial expression. For E. coli the outer membrane protein intimidin was chosen as an anchor for the antibody libraries.7
The next level of complexity - being closer to natural conditions – is the display of full length antibodies on mammalian cells.8 For this purpose, Chinese hamster ovary (CHO) cells with a single genome integration site were chosen to avoid the display of multiple different antibodies on a single cell. The heavy chain fragment was fused to the platelet-derived growth factor receptor (PDGFR) to establish an anchor at the cell membrane to which the respective light chains could assemble. The obvious advantages of that system are the ability to mirror the avidity effects of full length antibodies with two identical binding sites and the normal processing in mammalian host cells, giving a first indication of manufacturability in a production environment.
In vivo screening and selection
All the methods described above depend on the artificial construction of antibody libraries and their in vitro screening and selection. This approach has certainly proven highly successful and very efficient, but lacks elements of the natural immune system effects, such as immunoglobulin DNA re-arrangement, somatic recombination, isotype switching and affinity maturation by somatic hypermutation (SHM), which happens typically after immunisation of animals. Therefore, scientists have searched for methods to maintain the natural generation of antibodies, but that result in human variants. One solution is the grafting and replacement of complete mouse genes with human genes, resulting in the so-called Xenomouse.9 Immunising these transgenic mice with human antigens delivers fully human antibodies with high affinity. As the human constant region has the potential to not function optimally in a mouse context, another transgenic variant, the Velocimouse, was established wherein only the variable regions were replaced, requiring a subsequent in situ humanisation of the constant region.10 In the same year a comparable transgenic mouse was established by a different group.11 Besides mice, transgenic rats are also used for antibody generation.12
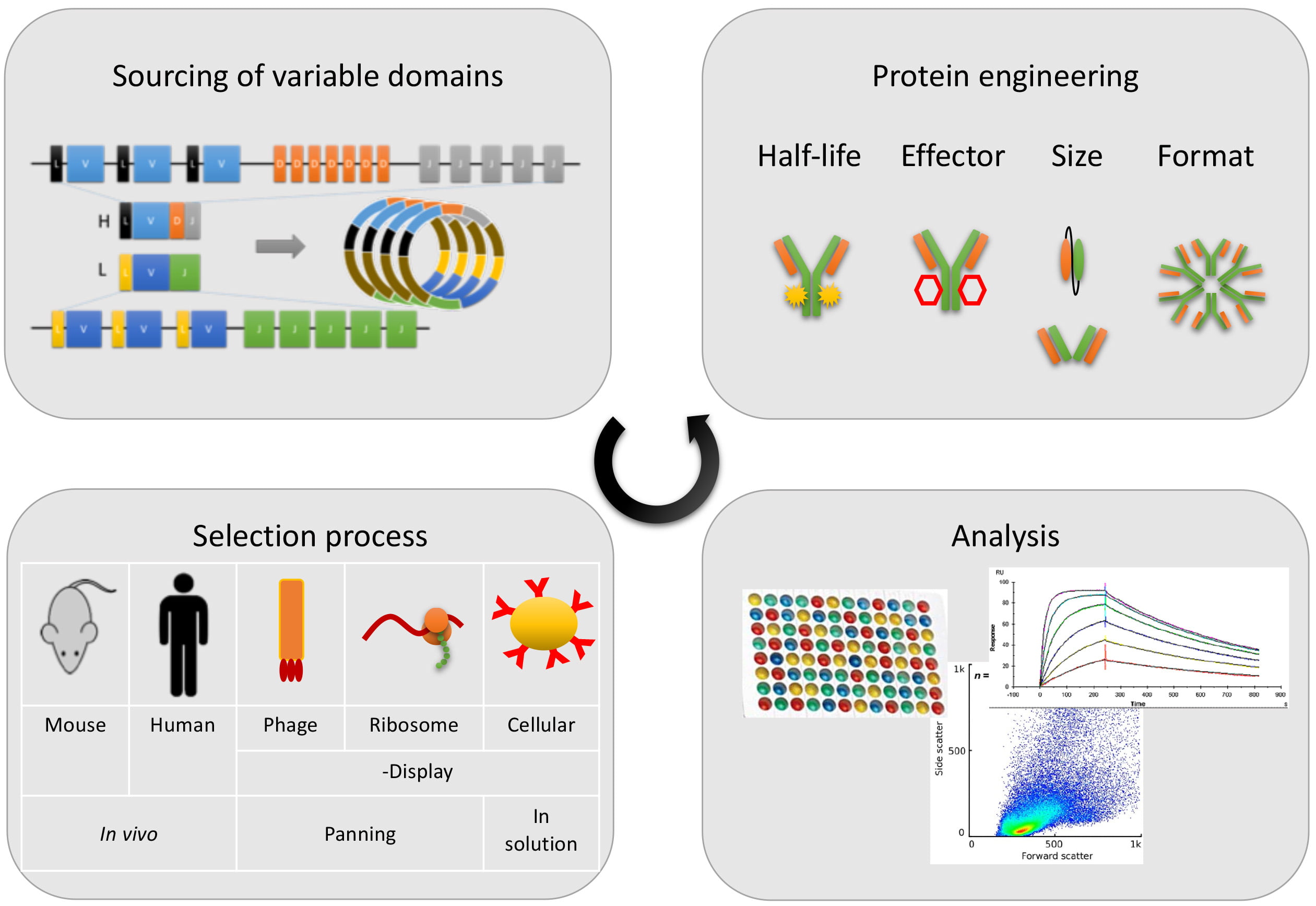
Figure 1: Typical antibody discovery steps
Another interesting source for human antibodies includes the isolation of B-cells from humans who are exposed to pathogens or following vaccination. One striking example is neutralising antibodies isolated from patients infected by viruses. Peripheral blood memory B-cells are collected and immortalised by transfection with Ebstein Barr Virus (EBV) and screened for antibody binding.13 Alternatively, fusion with human myeloma cells to obtain fully human hybridomas can be performed.14 Even simpler is the direct isolation of B-cell clones from peripheral blood by single cell sorting.15 B-cell pools from healthy volunteers can also be used for ex vivo immunisation in the presence of T-cells and subsequent immortalisation.12 In table 1 standard technologies are compared.
Antibody engineering
Initially, researchers were satisfied having obtained high affinity IgG molecules that could be expressed above 1g/L in clonal cell lines. However, in many cases the therapeutic effect was limited – either due to insufficient pharmacokinetics or by the lack of other important effector functions that were not screened from the beginning. These disadvantages are eliminated by protein engineering to modify antibody sequences for the desired functionalities, primarily located in the constant region. For instance, plasma half-life is highly dependent on the binding to the neonatal Fc gamma receptor (FcRn) and recycling through the endosome. The mutagenesis of two critical amino acids T250Q/M428L increases the half-life up to 11 fold.17
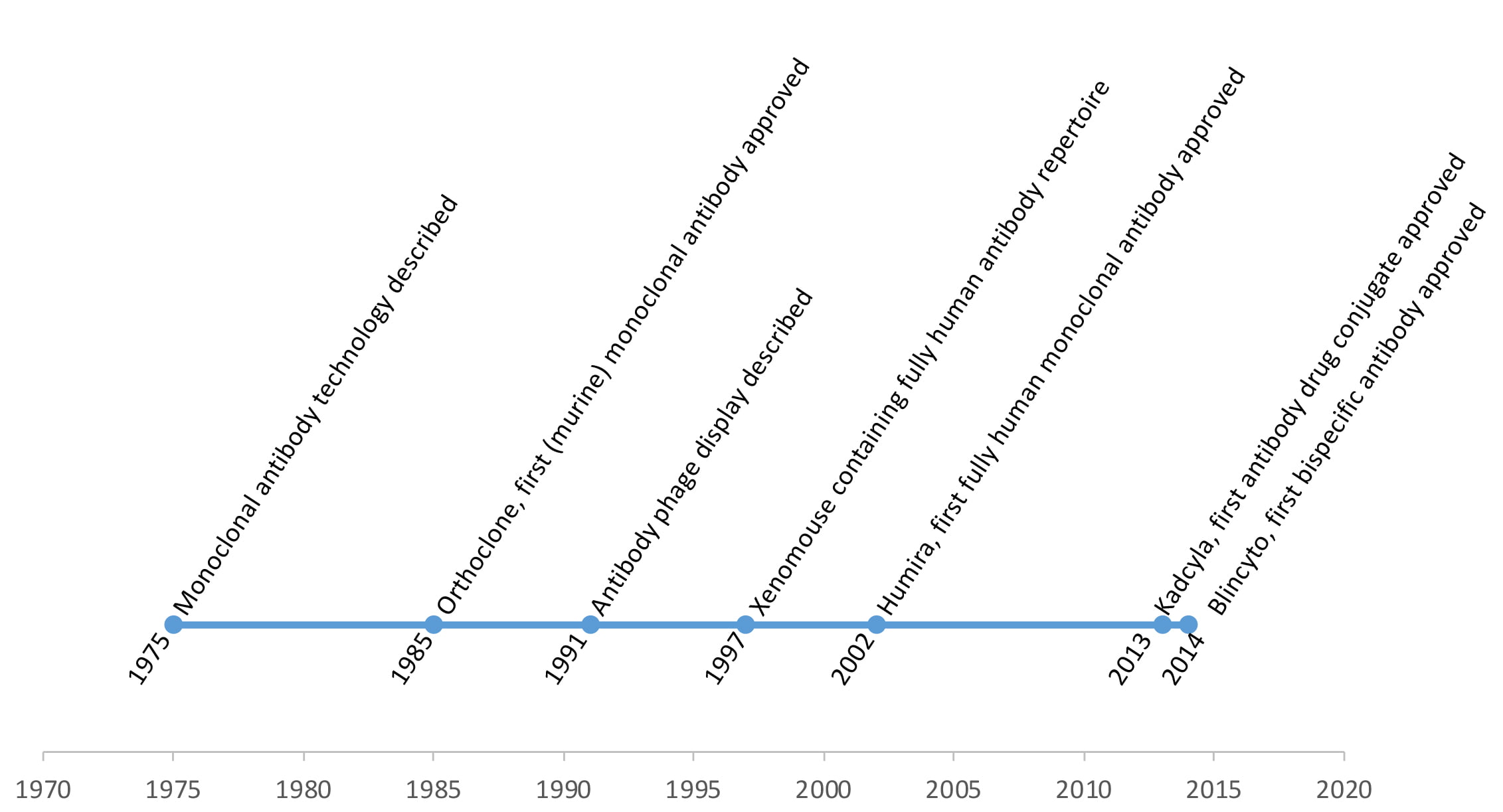
Figure 2: Milestones of antibody discovery
Other receptor functions like antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC) can be improved by glycoengineering.18 If effector functions are neglected, the functionality of the antibody can be reduced to its variable domain in the form of a scFv. Other fragment formats include the F(ab’)2 with two variable arms but no Fc domain or small bispecific antibody derivatives. There are currently more than 60 different bispecific construction designs described in the literature.2 Aside from bispecific functionalities, multimerisation with far more binding sites and polyvalence have been developed: a very recent example being hexameric antibodies that can induce complement activation.19 Another variant is the polymeric IgG that is joined by a 16 amino acid peptide from the tail of IgM fused to carboxyl terminus of the IgG.20 Looking at the different sources of variable domains currently used, it is quite striking that a wide variety is derived from rodents, humans and even species like camels; with some researchers evaluating the potential of sharks and chickens as well.21
Conclusion
Both in vitro and in vivo screening approaches have their merits, and currently antibodies derived from both techniques have been therapeutically successful. Figure 1 displays the four typical steps. The deeper understanding of sequence and structure-based functionality has led to the significant improvements of antibody efficacies through protein engineering. Nowadays, antibodies do not necessarily have to look like conventional IgG molecules, as widespread multimerisation and miniaturisation designs have been established. The next frontier might be to address different targets within the cell in place of the usual surface-bound or secreted molecules. Antibodies as natural molecules are still the most important and versatile class of large biological molecules to fight a wide range of diseases, and tremendous progress has been achieved over the last 40 years. Figure 2 summarises the key milestones in a timeline. It will be exciting to see how this success story continues.
Biography
Dr. Stefan R. Schmidt MBA is the Managing Director of s4advice, a biopharma consulting company, founded in 2011. His activities focus on CMC strategy, workflow optimization, fundraising, facility expansion, CAPEX projects and business development. In his career of more than 20 years he held several senior executive positions in international pharma and biotech companies.
References
- Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–7.
- Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. 2018;9(1):86–120.
- Verhoeyen M, Milstein C, Winter G. Reshaping human antibodies: Grafting an antilysozyme activity. Science. 1988;239(4847):1534–6.
- Barbas CF, Kang AS, Lerner RA, Benkovic SJ. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc Natl Acad Sci. 1991;88(18):7978–82.
- Hanes J, Pluckthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci. 1997;94(10):4937–42.
- Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15(6):553–7.
- Salema V, Fernández LÁ. Escherichia coli surface display for the selection of nanobodies. Microb Biotechnol. 2017;10(6):1468–84.
- Zhou C, Jacobsen FW, Cai L, Chen Q, Shen WD. Development of a novel mammalian cell surface antibody display platform. MAbs. 2010;2(5):508–18.
- Mendez MJ, Green LL, Corvalan JRF, Jia XC, Yang XD, Gallo ML, et al. Functional transplant of megabase human immunoglobulin loci recapitulates human antibody response in mice. Nat Genet. 1997;15(2):146–56.
- Murphy AJ, Macdonald LE, Stevens S, Karow M, Dore AT, Pobursky K, et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc Natl Acad Sci. 2014;111(14):5153–8.
- Lee EC, Liang Q, Ali H, Bayliss L, Beasley A, Bloomfield-Gerdes T, et al. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat Biotechnol. 2014;32(4):356–63.
- Osborn MJ, Ma B, Avis S, Binnie A, Dilley J, Yang X, et al. High-Affinity IgG Antibodies Develop Naturally in Ig-Knockout Rats Carrying Germline Human IgH/Ig /Ig Loci Bearing the Rat CH Region. J Immunol. 2013;190(4):1481–90.
- Wang H, Ma C, Lu Y, Ji X, Pang Y, Hua F, et al. Generation of human neutralizing monoclonal antibodies against the 2009 pandemic H1N1 virus from peripheral blood memory B lymphocytes. Cell Mol Immunol. 2013;10(5):403–12.
- Smith SA, Crowe JE. Use of Human Hybridoma Technology To Isolate Human Monoclonal Antibodies. Microbiol Spectr. 2015;3(1):1–12.
- Pinder CL, Kratochvil S, Cizmeci D, Muir L, Guo Y. Isolation and Characterisation of Antigen-Specific Plasmablasts Using a Novel Flow Cytometry-Based Immunoglobulin Capture Assay ( ICA ). J Immunol. 2018;199(12):4180–8.
- Li J, Sai T, Berger M, Chao Q, Davidson D, Deshmukh G, et al. Human antibodies for immunotherapy development generated via a human B cell hybridoma technology. Proc Natl Acad Sci U S A. 2006;103(10):3557–62.
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IWL, Sproule TJ, et al. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28(2):157–9.
- Wang Q, Chung CY, Chough S, Betenbaugh MJ. Antibody glycoengineering strategies in mammalian cells. Vol. 115, Biotechnology and Bioengineering. 2018. p. 1378–93.
- Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343:1260–3.
- Mekhaiel DNA, Czajkowsky DM, Andersen JT, Shi J, El-faham M, Doenhoff M, et al. Polymeric human Fc-fusion proteins with modified effector functions. Sci Rep. 2011;1:1–11.
- Strohl W. Antibody Discovery: Sourcing of Monoclonal Antibody Variable Domains. Curr Drug Discov Technol. 2014;11(1):3–19.
The rest of this content is restricted - login or subscribe free to access
 Thank you for visiting our website. To access this content in full you'll need to login. It's completely free to subscribe, and in less than a minute you can continue reading. If you've already subscribed, great - just login.
Thank you for visiting our website. To access this content in full you'll need to login. It's completely free to subscribe, and in less than a minute you can continue reading. If you've already subscribed, great - just login.
Why subscribe? Join our growing community of thousands of industry professionals and gain access to:
- quarterly issues in print and/or digital format
- case studies, whitepapers, webinars and industry-leading content
- breaking news and features
- our extensive online archive of thousands of articles and years of past issues
- ...And it's all free!
Click here to Subscribe today Login here
Related organisations
s4advice - Gunzenhausen - Germany