

Targeting the ubiquitin-proteasome system
Posted: 11 May 2014 | Kamyar Hadian (HelmholtzZentrum Munchen), Kenji Schorpp (HelmholtzZentrum Munchen) | No comments yet
The ubiquitin-proteasome system (UPS) is a hierarchical enzymatic cascade that plays an essential role in a broad range of cellular pathways and is found in cytoplasmic and nuclear actions of all eukaryotic cells…

Ubiquitination of cellular target proteins has been implicated in protein degradation, signal transduction, cell cycle regulation and response to DNA damage. The emerging knowledge of the molecular mechanisms and correlation of deregulation of the ubiquitin system in human diseases, including cancer, autoimmune diseases and neurodegenerative disorders, is uncovering new opportunities for therapeutic intervention. The first inhibitor targeting the UPS approved for clinical use was the proteasome inhibitor Bortezomib. The success story of Bortezomib has led to considerable interest in developing more inhibitors modulating the enzymes of the ubiquitin system.
The ubiquitin cascade
Ubiquitination is a posttranslational protein modification and involves the action of an enzyme cascade including the enzymes E1, E2 and E3 for the covalent attachment of ubiquitin to target proteins (Figure 1). The E1 enzyme activates and binds ubiquitin in an ATP-dependent manner1. In a second step, the E1 activating enzyme transfers ubiquitin to a cystein of an E2 conjugating enzyme. Then E3 ligases catalyse the attachment of the C-terminal glycin of ubiquitin to the ε-amino group of lysine residues or to the N-terminus of target proteins. This last step plays a crucial role in the recognition of targeted proteins and can occur via RING or HECT family E3 ligases. RING-type E3 ligases bind the E2-ubiquitin and the target protein in order to facilitate the transfer2. The RING E3 ligases are further sub-divided into the monomeric enzymes and the multimeric ligase complexes, which include the SCF family of E3 ligases. HECT ligases contain an active cystein that forms a thioester bond with the C-terminus of ubiquitin before transferring it to the protein substrate3. Repeated cycles of the E1, E2 and E3 enzyme actions allow the conjugation of polyubiquitin chains, which are important for the regulation of many cellular functions (Figure 1).
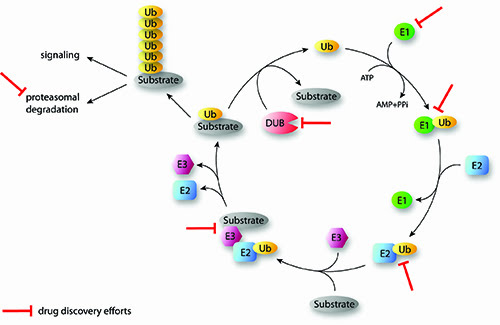
FIGURE 1: Overview of the sequential actions of the ubiquitination cascade and examples of drug discovery efforts (Ubiquitination of substrates involves the activity of three enzymes (E1, E2, E3) working in the depicted order. DUBs cleave ubiquitin from substrates and reverse the ubiquitination process. Iterative cycles will generate polyubiquitinated proteins that dependent on the generated type of chain linkage can be targeted to proteasomal degradation or may have signaling functions. Some points of drug discovery intervention are indicated. Ub: Ubiquitin; DUB: Deubiquinating enzyme; E1: Activating enzyme; E2: Conjugating enzyme; E3: Ligating enzyme).
Ubiquitin linkages and ubiquitin binding domains (UBDs)
Ubiquitination was initially discovered as a signal to mark proteins for subsequent degradation by the 26S-Proteasome4. However, there are a manifold of cellular functions that are regulated by nonproteolytic ubiquitination including cell cycle control, endocytosis, DNA repair and signal transduction5. The diverse functions of protein-ubiquitination are dependent on the position of the attached ubiquitin on the target protein and whether the target is monoubiquitinated at one or multiple sides or is decorated with polyubiquitin moieties. Ubiquitin chain linkages can be formed via seven lysine residues (K6, K11, K27, K29, K33, K48 and K63) within ubiquitin or via the N-terminal methionine (M1) of ubiquitin leading to head-to-tail linkage of ubiquitin6. The covalent attachment of polyubiquitin molecules linked via K48 marks proteins for degradation by the 26S-Proteasome, whereas one of the best characterised ubiquitin chain types involved in cell signalling events, such as receptor signalling or DNA damage response, is the K63 linked ubiquitin chain7. The N-terminal M1-linked linear polyubiquitination participates alongside with K63 chains in the regulation of the canonical NF-κB signalling8. In conjunction with noncovalent binding partners containing UBDs to recognise the different ubiquitin chains, the fate of the ubiquitinated proteins and the underlying pathway is regulated.
Drug Target Review has just announced the launch of its NEW and EXCLUSIVE report examining the evolution of AI and informatics in drug discovery and development.
In this 63 page in-depth report, experts and researchers explore the key benefits of AI and informatics processes, reveal where the challenges lie for the implementation of AI and how they see the use of these technologies streamlining workflows in the future.
Also featured are exclusive interviews with leading scientists from AstraZeneca, Auransa, PolarisQB and Chalmers University of Technology.
Deubiquitinating enzymes (DUBs)
Like acetylation and phosphorylation, ubiquitination is a reversible process. Ubiquitin moieties can be removed from target proteins by DUBs (Figure 1). Thus, DUBs can prevent protein degradation, revert signalling processes and contribute to cell cycle control and protein homeostasis9. The human genome encodes approximately 100 DUBs that can be classified into five subfamilies, i.e. ubiquitin specific proteases (USP), ubiquitin Cterminal hydrolyses (UCH), Josephine domain proteases, ovarian tumour proteases (OTU) and JAB1/MPN/MOV34 (JAMM) metallo-enzyme proteases10. It is suggested that DUBs represent good drug candidates since their deregulation has been connected to certain diseases11. Moreover, some DUB enzymes show target and linkage specific activity and contain well characterised catalytic pockets6. For instance, most of the OTU domain DUBs show intrinsic linkage specificity in vitro and many of them are crystallised in complex with ubiquitin chains12.
Ubiquitin like proteins (UBL)
Several protein modifiers related to the ubiquitin system were discovered during the past few decades. Ubiquitin like proteins (UBLs) such as SUMO, Nedd8 or ISG15 are members of the eukaryotic ubiquitin family and share the ubiquitin fold and similar mechanisms of activation. They regulate a diverse set of cellular processes, including nucleus-cytoplasmic shuttling, autophagy, translation, and antiviral pathways. Approximately 20 UBLs are known to be involved in the posttranslational modification of various proteins and other macromolecules13. Thus, the identification of UBLs adds another layer of complexity and creates a large pool of potential novel drug targets.
Proteasome
The 26S-proteasome is a cylindrical cellular organelle that recognises and degrades K48-polyubiquitinated proteins. It consists of one core subunit (20S unit) flanked by two regulatory subunits (19S unit) 14. In 2003, Bortezomib was the first drug targeting a component of the ubiquitin family approved by the FDA4. The clinical success of Bortezomib was surprising, because the consequence of blocking protein degradation and by that fostering stabilisation and accumulation of ubiquitinated proteins appeared to be a toxic event for any cell. However, Bortezomib leads to substantial but manageable toxicity in patients treated against multiple myeloma and mantle cell lymphomas.
In summary, the ubiquitin-proteasome system is a multistep process with all enzymes involved in regulation of this process being potential targets for therapeutic intervention. Additionally, the ubiquitin like proteins together with all specific binding partners to the posttranslational modifications create a manifold pool of drug targets, which are discussed in this review.
Small molecules targeting the UPS
Two ubiquitin E1 enzymes, UBA1 and UBA6, have been identified to date15. Targeting the E1 enzyme impairs the whole enzymatic ubiquitin cascade and substrate regulation. Consequently, E1 inhibitors act unspecific rather than influencing one specific ubiquitin substrate. The identification and unanticipated success of proteasome inhibitors changed the way of thinking that also targeting E1 might be rewarding. Efforts to screen for general inhibitors of the ubiquitin enzymatic machinery led to the identification of Pyr41 an E1 enzyme inhibitor. Pyr41 is a cell permeable inhibitor and may block the catalytic cysteine of ubiquitin E1 enzymes16. An example for a success story targeting E1 enzymes originates from the small molecule MLN4924. MLN4924 attacks the charged E1 of the UBL Nedd8 and forms a covalent adduct with Nedd8. It was shown that treatment of several tumours with MLN4924 inhibits growth and this molecule is currently in clinical studies17,18.
The next layer of the ubiquitination cascade implies the activity of E2 conjugation enzymes. Approximately 30 E2 enzymes are known in human. Due to the greater number of E2 enzymes, targeting E2 enzymes is a more specific interference when compared to E1 enzymes. There are theoretically four different ways to inhibit the ubiquitin transfer via E2s. A small molecule could: (i) block the E1-E2 interaction; (ii) target and inactivate the active side E2 cystein; (iii) block the E2-E3 interaction interface; or (iv) act as an allosteric inhibitor. In 2011, Ceccarelli and colleagues reported an allosteric inhibitor (CC0651) for the E2 enzyme Cdc34. It was shown that CC0651 does not impair Cdc34 interactions with E1 or E3 enzymes or influences the formation of the ubiquitin thioester but instead interferes with the discharge of ubiquitin to acceptor lysine residues. The authors could also present data that treatment of cancer cells with CC0651 can inhibit proliferation19.
Selective inhibition of E3 ligases is likely to be much more specific than targeting E1 and E2 enzymes or the proteasome. Around 600 E3 ligases are known and mutations or uncontrolled activity of them are often associated with human disease20,21. RING E3 ligases mediate the interaction between E2 enzymes charged with ubiquitin and the substrate to be ubiquitinated.
The anti-apoptotic proteins (IAPs) E3-RING ligases are actively investigated by many companies and tested for therapeutic intervention. Several approaches were used, including the screening of peptide or peptidomimetic libraries to identify analogues of Smac22,23. Smac is a naturally occurring protein that binds IAPs and thereby triggers cell death. Studies with Smac mimetics demonstrated that these mimetic molecules are very promising in promoting TNFα-dependent apoptosis in cancer cells24-26. Another example for interfering with E3s is the RING ligase HDM2 that targets the anti-tumour suppressor p53 for degradation. Inhibition of HDM2 can restore and elevate p53 expression in cancers containing wild-type p53, leading to apoptosis and growth inhibition27. Nutlin3 and JNJ26854165 are two HDM2 antagonising inhibitors in clinical trials28,29. Moreover, the small molecule RITA binds to the N-terminus of p53 and induces a conformational change blocking its interaction with HDM230,31. Thus, targeting both the surface of the E3 as well as the E3 substrate is a valid and promising strategy. Based on the studies, reporting a class of high-affinity HDM2 inhibitors represented by MI-219 led to the identification of the second generation HDM2 inhibitor, RG7388, with high potency and selectivity32. In summary, all three enzymes of the ubiquitination cascade (E1, E2 and E3) are in use for the development of small molecule drugs. However, the specificity and selectivity of the drug vary dependent on the particular enzyme being targeted and needs to be considered on a case-by-case basis when starting a new drug discovery project.
Besides targeting the E3 ligase HMD2, another option was highlighted by the development of small molecule inhibitors for deubiquitinating enzymes (DUBs). DUBs can remove ubiquitin moieties from the substrate and thus regulate the stability of proteins. It was demonstrated that abolishment of USP7 can lead to destabilisation of HDM2 and as a consequence stabilise p5333. Therefore companies have developed molecules to inhibit USP7 and by that to enhance polyubiquitination and subsequent degradation of HDM234. The example of p53 regulation reveals a basic phenomenon that either targeting the E3 ligase or a DUB enzyme can be valid strategies to influence homeostasis of a specific protein substrate. In the meantime, many researchers and companies are developing inhibitors for a broad set of DUBs. For instance, UCHL1 plays a role in neurodegenerative disease and cancer and is therefore of great therapeutic interest. However, so far the existing selective UCHL1 inhibitors show moderate potency and more work has to be done to enter clinical trials35,36.
Finally, a mode of action of the proteasome inhibitor Bortezomib might come along with the p53 protein as well. Treatment with Bortezomib stabilises p53 (beside many other proteins) and thus induces apoptosis in cancer cells. This seems to be a consequence of the described role for Bortezomib to bind the β1 and β5 subunit of the proteasome degradation chamber and thereby to inhibit the proteasome degradation activity37. In addition to Bortezomib, other experimental preclinical inhibitors have been developed that target the DUB activity of one of three DUBs associated with the 19S-proteasome particle38. In a very recent study, a new proteasome inhibitor (RA190) was published that targets the UBD protein RPN13 of the 19S-proteasome particle39. The p53 protein is probably the best investigated protein with regards to drug development of the UPS and is a good example to highlight the different options how to target this system at different stages of the enzymatic cascade. All the examples illustrate that the UPS, the UBLs and noncovalent interaction partners provide a rich source of attractive molecular targets for pharmacological intervention.
Perspective
Basic research data together with newfound biomarkers reveal novel associations of the ubiquitin system with human diseases. In particular, deregulation of E3 ligases and DUBs clearly contribute to cancerogenesis and are directly linked to oncogenic and tumour suppressor phenotypes11. For this reason, human E3 ligases or DUBs are considered as promising novel therapeutic targets11,40,41. Interfering with these protein classes might have the advantage to act more specifically on a subset of ubiquitination substrates and these drugs are therefore expected to be less toxic than global inhibitors of the ubiquitin system that target E1/E2 enzymes or the proteasome. Developing screening assays for E3 activity is conceptually more challenging than for DUBs. Some RING E3 complexes consist of more than 10 protein subunits and the catalytically active side of monomeric HECT E3 ligases is also not easily targetable. Enzymatic assays for E3 ligase activity require additional enzymes such as an E1 and an E2. The recent development of robust and functional assays that reconstitute entire E3 ligase cascades drastically improved the examination of inhibitors for E3 ligases in HTS format. However, more development on these types of assays may further facilitate activities on E3 drug development. So far, most attempts have been done to identify inhibitors of the E3-substrate complex and little effort has been devoted to interrupt the E2-E3 interaction. It has been reported that E2-E3 interactions are usually weak and therefore targeting this protein-protein interaction has a great potential for future small molecule screening campaigns42. In contrast to E3 ligases the assay development for DUBs is much easier since most of the assays include only two components, i.e. the enzyme and ubiquitin-linkage substrate, respectively. In addition, DUBs can be readily profiled using ubiquitin-based chemical probes targeting the proteolytic catalysis. A recent study published in Science illustrates a new method using ubiquitin variants for the modulation of USPs43. Moreover, active site molecular probes on the basis of the ubiquitin scaffold labelled with fluorophores are also very useful for the characterisation of DUBs under physiological conditions44.
Together with state-of-the-art proteomics, these methods enable cellular validation of the specificity of small molecule DUB inhibitors and expand the understanding of DUB enzymatic activity. Increasing knowledge will help to design chemical compound libraries that target the different DUB families more specifically and might help to reduce attrition rate at later stages of drug discovery processes. In general, many new ideas and approaches will arise from basic research to specify strategies for targeting the UPS. Thus, considering the high diversity of ubiquitin and UBL functions it is very likely that ubiquitin drug discovery will create a large pool of substrates for the treatment of various diseases.
Biographies
 KENJI SCHORPP is a research scientist at the Assay Development and Screening Platform, Helmholtz-Zentrum München, Munich, Germany. He is responsible for assay development and screening of early drug discovery projects and his scientific focus is drug discovery in the ubiquitin system. Prior to this, he worked as a Postdoc and PhD at the Max-Planck-Institute of Biochemistry where he investigated the regulation and function of ubiquitin and ubiquitin like proteins.
KENJI SCHORPP is a research scientist at the Assay Development and Screening Platform, Helmholtz-Zentrum München, Munich, Germany. He is responsible for assay development and screening of early drug discovery projects and his scientific focus is drug discovery in the ubiquitin system. Prior to this, he worked as a Postdoc and PhD at the Max-Planck-Institute of Biochemistry where he investigated the regulation and function of ubiquitin and ubiquitin like proteins.
 KAMYAR HADIAN is the head of the Assay Development and Screening Platform at the Helmholtz Zentrum München in Munich, Germany. He studied biology at the Technical University of Munich (TUM) and gained his PhD at the Helmholtz Zentrum München/Ludwig Maximilians University in the field of Virology (HIV). After a short PostDoc period in the field of assay development and screening, he was appointed the head of Assay Development and Screening Platform at the Helmholtz Zentrum München. He has experience in designing biochemical as well as cell-based assays and to carry out HTS or High-Content Screening. His scientific focus is finding novel therapeutics in the ubiquitin system.
KAMYAR HADIAN is the head of the Assay Development and Screening Platform at the Helmholtz Zentrum München in Munich, Germany. He studied biology at the Technical University of Munich (TUM) and gained his PhD at the Helmholtz Zentrum München/Ludwig Maximilians University in the field of Virology (HIV). After a short PostDoc period in the field of assay development and screening, he was appointed the head of Assay Development and Screening Platform at the Helmholtz Zentrum München. He has experience in designing biochemical as well as cell-based assays and to carry out HTS or High-Content Screening. His scientific focus is finding novel therapeutics in the ubiquitin system.
References
- L. Haas, J. V. Warms, A. Hershko and I. A. Rose (1982) Ubiquitin-activating enzyme. Mechanism and role in protein-ubiquitin conjugation. J Biol Chem 257:2543-2548
- Ozkan, H. Yu and J. Deisenhofer (2005) Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. Proc Natl Acad Sci U S A 102:18890-18895
- Rotin and S. Kumar (2009) Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol 10:398-409
- Adams (2002) Development of the proteasome inhibitor PS-341. Oncologist 7:9-16
- Kerscher, R. Felberbaum and M. Hochstrasser (2006) Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol 22:159-180
- Komander and M. Rape (2012) The ubiquitin code. Annu Rev Biochem 81:203-229
- Hoege, B. Pfander, G. L. Moldovan, G. Pyrowolakis and S. Jentsch (2002) RAD6- dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419:135-141
- Iwai (2011) Linear polyubiquitin chains: a new modifier involved in NFkappaB activation and chronic inflammation, including dermatitis. Cell Cycle 10:3095-3104
- Song and M. Rape (2008) Reverse the curse–the role of deubiquitination in cell cycle control. Curr Opin Cell Biol 20:156-163
- Komander, M. J. Clague and S. Urbe (2009) Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 10:550-563
- Shi and S. R. Grossman (2010) Ubiquitin becomes ubiquitous in cancer: emerging roles of ubiquitin ligases and deubiquitinases in tumorigenesis and as therapeutic targets. Cancer Biol Ther 10:737-747
- E. Mevissen, M. K. Hospenthal, P. P. Geurink, P. R. Elliott, M. Akutsu, N. Arnaudo, R. Ekkebus, Y. Kulathu, T. Wauer, F. El Oualid, S. M. Freund, H. Ovaa and D. Komander (2013) OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 154:169-184
- G. van der Veen and H. L. Ploegh (2012) Ubiquitin-like proteins. Annu Rev Biochem 81:323-357
- Finley (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem 78:477-513
- Jin, X. Li, S. P. Gygi and J. W. Harper (2007) Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 447:1135-1138
- Yang, J. Kitagaki, R. M. Dai, Y. C. Tsai, K. L. Lorick, R. L. Ludwig, S. A. Pierre, J. P. Jensen, I. V. Davydov, P. Oberoi, C. C. Li, J. H. Kenten, J. A. Beutler, K. H. Vousden and A. M. Weissman (2007) Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res 67:9472-9481
- A. Soucy, P. G. Smith, M. A. Milhollen, A. J. Berger, J. M. Gavin, S. Adhikari, J. E. Brownell, K. E. Burke, D. P. Cardin, S. Critchley, C. A. Cullis, A. Doucette, J. J. Garnsey, J. L. Gaulin, R. E. Gershman, A. R. Lublinsky, A. McDonald, H. Mizutani, U. Narayanan, E. J. Olhava, S. Peluso, M. Rezaei, M. D. Sintchak, T. Talreja, M. P. Thomas, T. Traore, S. Vyskocil, G. S. Weatherhead, J. Yu, J. Zhang, L. R. Dick, C. F. Claiborne, M. Rolfe, J. B. Bolen and S. P. Langston (2009) An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458:732-736
- E. Brownell, M. D. Sintchak, J. M. Gavin, H. Liao, F. J. Bruzzese, N. J. Bump, T. A. Soucy, M. A. Milhollen, X. Yang, A. L. Burkhardt, J. Ma, H. K. Loke, T. Lingaraj, D. Wu, K. B. Hamman, J. J. Spelman, C. A. Cullis, S. P. Langston, S. Vyskocil, T. B. Sells, W. D. Mallender, I. Visiers, P. Li, C. F. Claiborne, M. Rolfe, J. B. Bolen and L. R. Dick (2010) Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell 37:102- 111
- F. Ceccarelli, X. Tang, B. Pelletier, S. Orlicky, W. Xie, V. Plantevin, D. Neculai, Y. C. Chou, A. Ogunjimi, A. Al-Hakim, X. Varelas, J. Koszela, G. A. Wasney, M. Vedadi, S. Dhe-Paganon, S. Cox, S. Xu, A. Lopez-Girona, F. Mercurio, J. Wrana, D. Durocher, S. Meloche, D. R. Webb, M. Tyers and F. Sicheri (2011) An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell 145:1075-1087
- S. Fakharzadeh, S. P. Trusko and D. L. George (1991) Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J 10:1565-1569
- L. Welcsh and M. C. King (2001) BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet 10:705-713
- K. Oost, C. Sun, R. C. Armstrong, A. S. Al-Assaad, S. F. Betz, T. L. Deckwerth, H. Ding, S. W. Elmore, R. P. Meadows, E. T. Olejniczak, A. Oleksijew, T. Oltersdorf, S. H. Rosenberg, A. R. Shoemaker, K. J. Tomaselli, H. Zou and S. W. Fesik (2004) Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem 47:4417-4426.
- Sun, Z. Nikolovska-Coleska, C. Y. Yang, L. Xu, M. Liu, Y. Tomita, H. Pan, Y. Yoshioka, K. Krajewski, P. P. Roller and S. Wang (2004) Structure-based design of potent, conformationally constrained Smac mimetics. J Am Chem Soc 126:16686-16687
- Wu, J. Chai, T. L. Suber, J. W. Wu, C. Du, X. Wang and Y. Shi (2000) Structural basis of IAP recognition by Smac/DIABLO. Nature 408:1008-1012
- Varfolomeev, J. W. Blankenship, S. M. Wayson, A. V. Fedorova, N. Kayagaki, P. Garg, K. Zobel, J. N. Dynek, L. O. Elliott, H. J. Wallweber, J. A. Flygare, W. J. Fairbrother, K. Deshayes, V. M. Dixit and D. Vucic (2007) IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 131:669-681
- E. Vince, W. W. Wong, N. Khan, R. Feltham, D. Chau, A. U. Ahmed, C. A. Benetatos, S. K. Chunduru, S. M. Condon, M. McKinlay, R. Brink, M. Leverkus, V. Tergaonkar, P. Schneider, B. A. Callus, F. Koentgen, D. L. Vaux and J. Silke (2007) IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 131:682-693
- Wasylyk, R. Salvi, M. Argentini, C. Dureuil, I. Delumeau, J. Abecassis, L. Debussche and B. Wasylyk (1999) p53 mediated death of cells overexpressing MDM2 by an inhibitor of MDM2 interaction with p53. Oncogene 18:1921-1934
- Kojima, J. K. Burks, J. Arts and M. Andreeff (2010) The novel tryptamine derivative JNJ-26854165 induces wild-type p53- and E2F1-mediated apoptosis in acute myeloid and lymphoid leukemias. Mol Cancer Ther 9:2545-2557
- T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303:844-848
- A. Doggrell (2005) RITA–a small-molecule anticancer drug that targets p53. Expert Opin Investig Drugs 14:739-742
- Issaeva, P. Bozko, M. Enge, M. Protopopova, L. G. Verhoef, M. Masucci, A. Pramanik and G. Selivanova (2004) Small molecule RITA binds to p53, blocks p53- HDM-2 interaction and activates p53 function in tumors. Nat Med 10:1321-1328
- Ding, Z. Zhang, J. J. Liu, N. Jiang, J. Zhang, T. M. Ross, X. J. Chu, D. Bartkovitz, F. Podlaski, C. Janson, C. Tovar, Z. M. Filipovic, B. Higgins, K. Glenn, K. Packman, L. T. Vassilev and B. Graves (2013) Discovery of RG7388, a Potent and Selective p53-MDM2 Inhibitor in Clinical Development. J Med Chem, DOI: 10.1021/jm400487c
- M. Cummins, C. Rago, M. Kohli, K. W. Kinzler, C. Lengauer and B. Vogelstein (2004) Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 428:1 p following 486
- Colland, E. Formstecher, X. Jacq, C. Reverdy, C. Planquette, S. Conrath, V. Trouplin, J. Bianchi, V. N. Aushev, J. Camonis, A. Calabrese, C. Borg-Capra, W. Sippl, V. Collura, G. Boissy, J. C. Rain, P. Guedat, R. Delansorne and L. Daviet (2009) Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther 8:2286-2295
- H. Jara, D. D. Frank and P. H. Ozdinler (2013) Could dysregulation of UPS be a common underlying mechanism for cancer and neurodegeneration? Lessons from UCHL1. Cell Biochem Biophys 67:45-53
- H. Mermerian, A. Case, R. L. Stein and G. D. Cuny (2007) Structure-activity relationship, kinetic mechanism, and selectivity for a new class of ubiquitin C-terminal hydrolase-L1 (UCH-L1) inhibitors. Bioorg Med Chem Lett 17:3729-3732
- Altun, P. J. Galardy, R. Shringarpure, T. Hideshima, R. LeBlanc, K. C. Anderson, H. L. Ploegh and B. M. Kessler (2005) Effects of PS-341 on the activity and composition of proteasomes in multiple myeloma cells. Cancer Res 65:7896-7901
- H. Lee, M. J. Lee, S. Park, D. C. Oh, S. Elsasser, P. C. Chen, C. Gartner, N. Dimova, J. Hanna, S. P. Gygi, S. M. Wilson, R. W. King and D. Finley (2010) Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467:179-184
- K. Anchoori, B. Karanam, S. Peng, J. W. Wang, R. Jiang, T. Tanno, R. Z. Orlowski, W. Matsui, M. Zhao, M. A. Rudek, C. Hung, X. DChen, K. J. Walters and R. B. S. Roden (2013) A bis-Benzylidine Piperidone Targeting Proteasome Ubiquitin Receptor RPN13/ADRM1 as a Therapy for Cancer. Cancer Cell 24:791-805
- Ciechanover, A. Orian and A. L. Schwartz (2000) The ubiquitin-mediated proteolytic pathway: mode of action and clinical implications. J Cell Biochem Suppl 34:40-51
- Nalepa, M. Rolfe and J. W. Harper (2006) Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov 5:596-613
- Ye and M. Rape (2009) Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol 10:755-764
- Ernst, G. Avvakumov, J. Tong, Y. Fan, Y. Zhao, P. Alberts, A. Persaud, J. R. Walker, A. M. Neculai, D. Neculai, A. Vorobyov, P. Garg, L. Beatty, P. K. Chan, Y. C. Juang, M. C. Landry, C. Yeh, E. Zeqiraj, K. Karamboulas, A. Allali-Hassani, M. Vedadi, M. Tyers, J. Moffat, F. Sicheri, L. Pelletier, D. Durocher, B. Raught, D. Rotin, J. Yang, M. F. Moran, S. Dhe-Paganon and S. S. Sidhu (2013) A strategy for modulation of enzymes in the ubiquitin system. Science 339:590-595
- B. Kramer, B. Nicholson, B. M. Kessler and M. Altun (2012) Detection of ubiquitinproteasome enzymatic activities in cells: application of activity-based probes to inhibitor development. Biochim Biophys Acta 1823:2029-2037
Related topics
Disease research, Enzymes, Neurosciences, Oncology, Therapeutics, Ubiquitin-proteasome system