

LC3 – Autophagy’s achilles’ heel?
Posted: 10 April 2015 | Joana R Costa (TRRC University College London), Robin Ketteler (TRRC University College London) | No comments yet
Autophagy is a cellular homeostatic process regulating protein turnover as well as eliciting a stress response to starvation, chemical agents and pathogen infection. It has been implicated in the initiation and progression of several diseases, including neuro-degenerative disorders, Crohn’s disease, infections and cancer…
It has been proposed that therapeutics targeting autophagy will provide benefits in such instances, but uncertainties remain over the most suitable molecular target for intervention in the autophagy pathway. We propose that LC3 and its family members present a druggable class of proteins with sufficient genetic evidence and structural tractability to enable the development of therapeutic agents that modulate autophagosome formation in multiple diseases.
The role of autophagy in disease can be traced to one of two things: it can either result from mutation of autophagy genes, which contributes to the development of the disease, or in other cases autophagy can be a response to molecular changes in disease such as DNA damage in cancer2. Some examples of autophagy gene mutations that contribute to disease include haplo-insufficiency of Beclin in cancer3, mutation of ATG16L1 in Crohn’s disease4, de novo mutations in WDR45 leading to static encephalopathy of childhood with neurodegeneration in adulthood5, and recessive mutations in EPG5 in Vinci syndrome6.
To date, close to 50 clinical trials have been reported to investigate the impact of autophagy inhibition using hydroxychloroquine (HCQ) or its derivatives, mTOR inhibitors or AKT inhibitors, usually in combination with chemotherapeutic agents (www.clinicaltrials.gov). Whether the effects of such treatments are indeed caused by modification of the autophagic pathway or alternative processes that are also targeted by such treatments is not clear. It appears that there have been no clinical trials conducted to target the core autophagy machinery directly.
Drug Target Review has just announced the launch of its NEW and EXCLUSIVE report examining the evolution of AI and informatics in drug discovery and development.
In this 63 page in-depth report, experts and researchers explore the key benefits of AI and informatics processes, reveal where the challenges lie for the implementation of AI and how they see the use of these technologies streamlining workflows in the future.
Also featured are exclusive interviews with leading scientists from AstraZeneca, Auransa, PolarisQB and Chalmers University of Technology.
Candidate genes in the autophagy machinery that have been proposed as potential drug targets focus on the enzymatic reactions catalysed by kinases (ULK1/2, vps34/PI3K III) and proteases (ATG4 family members). Recently, inhibitors for ULK17 and vps348 have been described, whicht hold promise for further development, but these inhibitors are at an early phase of development.
One problem in targeting autophagy is the so-called ‘double-edged sword’ hypothesis9, i.e., the inhibition or activation of autophagy may both be beneficial for therapeutic strategies. The choice of inhibitor or activator may be context-dependent10. For instance, in neuro-degenerative diseases it is thought that activation of autophagy may be beneficial to cope with oxidative stress, damaged mitochondria and/or clearance of protein aggregation11. On the other hand, the role of autophagy in promoting cancer may depend on a complex interplay of genetic mutations, active signaling pathways and the tumour microenvironment10,12. In general, there are two main strategies for autophagy modulation: one is to inhibit lysosomal function or prevent lysosomal fusion as was shown by use of the anti-malarial drug chloroquine and derivatives. The other is to enhance autophagic flux and delivery to the lysosome (effectively enhancing lysosomal function).
LC3 and ATG8-like family members
A key complex required for autophagosome maturation is the ATG4-ATG8 conjugation system. In mammalian cells, multiple ATG8 isoforms exist, including MAP1LC3A, B, B2, C, GABARAP, GABARAPL1 and GABARAPL2 (Figure 1).
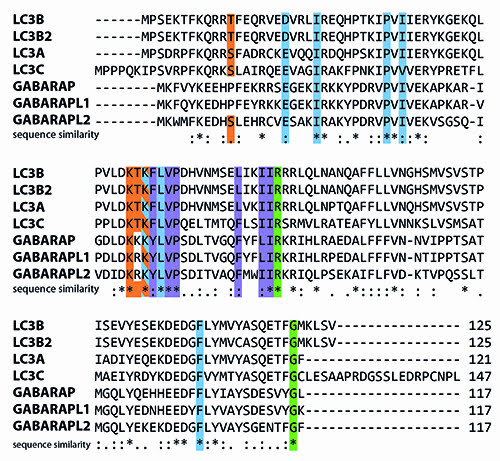
Figure 1: Human isoforms of LC3 aligned using the CLUSTAL W2 algorithm. LC3 has seven isoforms (Uniprot IDs): LC3A (Q9H492), LC3B (Q9GZQ8), LC3B2 (A6NCE7), LC3C (Q9BXW4), GABARAP (O95166), GABARAPL1 (Q9H0R8) and GABARAPL2 (P60520). Many of the important residues that have been identified are fully conserved. Residues highlighted in green are crucial for interaction with and cleavage by ATG4 proteases. Amino acids important for LIR interactions are displayed in blue and purple (α2 and ubiquitin-like domain hydrophobic pockets, respectively), and residues that have been shown to undergo post-translational modification (phosphorylation, (de-)acetylation) in at least one isoform are highlighted in orange.
These are differentially expressed in various tissues, thus suggesting a tissue-specific function13. Since members of the MAP1LC3 family are the best studied members in mammalian cells, we will use the term ‘LC3’ to collectively refer to these proteins. All isoforms are primarily processed from a pro-LC3 form into a LC3-I isoform by ATG4 family members, most notably ATG4B and to a lesser extent ATG4A. LC3-I is then converted to LC3-II by the addition of a phosphatidyl-ethanolamine anchor mediated by ATG3/7. This leads to incorporation of LC3-II into the inner and outer autophagosome membrane. Upon further maturation of the autophagosome, LC3-I is recycled by de-lipidation of LC3-II mediated by ATG4 members (Figure 2). Processing of LC3 is necessary for the complete formation of autophagosomes as suggested by studies performed in ATG4B null mouse embryonic fibroblasts (MEFs)14.
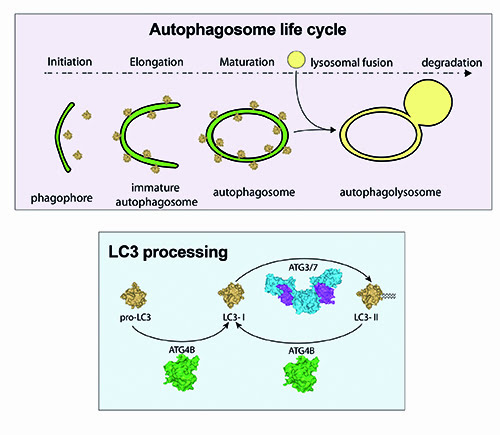
Figure 2: Schematic overview of LC3 processing. Pro-LC3 is converted into LC3-I by cleavage of an exposed glycine residue on the C-terminus, which is mediated by the cysteine protease ATG4B. ATG3/7 mediate the conversion of LC3-I to LC3-II by the addition of a phosphatidylethanolamine (PE) anchor leading to incorporation of LC3-II into autophagic membranes. Recycling of LC3-II to LC3-I is also mediated by ATG4B.
Most autophagy assays rely on measurement of expression or processing of the autophagy marker protein MAP1LC3B (LC3). These assays include fluorescence microscopy of LC3-positive cellular structures and LC3 isoform processing by western-blotting or indirect luciferase-based readouts15. While a lot of research has taken place on LC3 as a marker protein, the functional role as an essential component of the autophagosome maturation has not been conceptualised in the context of drug design.
LC3 in disease
LC3 proteins are key scaffold molecules bridging the autophagosome membrane with adapter molecules. Generally, there are two different functional types of interaction partners: one is the autophagy core components such as ATG5, 12, 16L1, which coat the autophagosome membrane and regulate its maturation; and secondly, selective receptors that bind to LC3 and deliver cargo destined for degradation to the autophago-lysosome. The latter process is referred to as selective autophagy and is active in disease-related cellular stresses such as mitochondrial damage (mitophagy) or defense against invading pathogens (xenophagy). In these cases, LC3 binds to autophagy receptor proteins such as p6216, NDP5217, NBR118 or optineurin19 which recognise ubiquitinated substrates. Most LC3 interactions are mediated by the LC3 interacting region (LIR) in the autophagy receptors binding to hydrophobic pockets in the LC3 protein (see figure 3B). Interestingly, LIR domain interactions can be highly specific. For instance, NDP52 selectively binds to LC3C, facilitating delivery of pathogens such as Salmonella enterica to the autophagosome and thus clearance20.
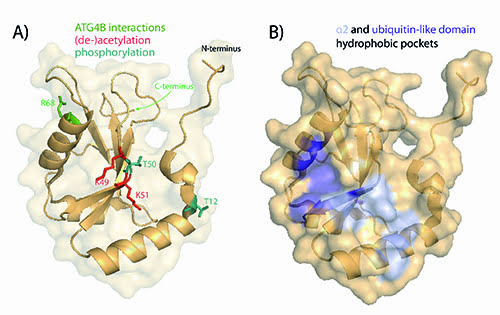
Figure 3: Structure of LC3B. The structures were generated with the PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC based on PDB: 3VTU38. A) Residues important for functional processing by ATG4B are shown in green and residues that have been shown to undergo post-translational modifications are displayed in teal (phosphorylation) and red (acetylation). B) Two hydrophobic pockets located by the α2 helix and the ubiquitin-like domain of LC3 that mediate interactions with LIR-containing proteins.
In the literature, LC3 expression levels have generally been equated to autophagy, since when LC3 expression has been found to be elevated in cells or tissues, it has been proposed that autophagy is more active. In agreement, LC3 has been proposed as being a diagnostic biomarker for several diseases including autophagic vacuolar myopathies21 and cancer22. However, the role of LC3 in disease is much more complex than merely being a marker for up-regulated autophagy. The expression levels of LC3 vary considerably amongst cancer types with high levels of basal autophagy, as measured by the number of LC3-autophagic vesicles in pancreatic cancer23 and gastrointestinal tumours24, and low levels in hypopharyngeal squamous cell carcinoma25. Furthermore, several studies have shown that high LC3B protein expression correlates with poor outcome in patient cells and shorter survival, pointing towards the use of LC3B as a prognostic marker in diseases such as astrocytoma and breast cancer26. Other studies have also suggested the involvement of other LC3 isoforms like MAP1LC3A in breast cancer development27. While it is tempting to correlate LC3 expression with autophagy activity, we propose that LC3 processing is a potential therapeutic target for modulation of the autophagy pathway in disease.
Targeting LC3 function
Targeting LC3 processing can be achieved by three possible routes, each of which is explored as follows.
Preventing C-terminal processing
Processing of pro-LC3 to LC3-I/II is a requirement for formation of the autophagosome. It can be envisioned that targeting this step with inhibitors will result in a block of autophagic vacuole (AV) formation. It might be possible to achieve this by compounds or peptides that bind to the C-terminus and thus prevent access to the ATG4 protease. Supporting this idea, it has been shown that the use of a catalytic inactive mutant of ATG4B sequesters the LC3 substrate, inhibiting its lipidation28. Accordingly, in the view of modulating the proteins that regulate LC3 function, the cysteine protease ATG4B is a main candidate. Modulation of ATG4 proteases has been a difficult task most likely due to their metabolic instability and lack of specificity of small-molecule compounds29. Nevertheless, high-throughput screening assays for monitoring ATG4B activity have been developed30 and efforts are underway to generate efficient ATG4 protease inhibitors.
Disrupting LC3 protein interactions
Targeting LC3 protein interactions can be beneficial to modulate specific autophagy complexes. Such modulation can be achieved by disruption of the LIR domain binding to hydrophobic pockets in LC3. In addition to selective autophagy receptor interactions, LIR domain interactions with LC3 occur in complexes that are potentially relevant to disease. For instance, the LIR domain in the ubiquitin ligase Cbl helps targeting of the protein kinase Src for autophagic degradation thereby promoting cancer cell survival31 and accordingly, disruption of such interactions may be a possible anti-cancer strategy. Furthermore, LIR domains mediate binding to autophagy core components such as ATG1 and it has been shown that mutations in the LIR domain of ATG1 can result in reduced autophagy32. Similarly, ATG4 family members and other core components of the autophagy machinery interact with LC3 via a LIR domain and it has been proposed that such interactions may be potential druggable targets33.
In addition to LIR domain interactions, there are potential other sites in LC3 such as the arginine-rich stretch R68-R70 of LC3 that has been shown to be required for ATG4B binding34. While targeting protein-protein interactions is generally not easily achieved, one can envisage small molecules or LIR peptides that disrupt the formation of these complexes. Interestingly, some LIR domain interactions are regulated by phosphorylation, presenting an alternative route for potential intervention.
Modulating post-translational modifications of LC3
LC3 itself underlies post-translational regulation (see figure 3A) and targeting these regulatory steps may result in an up- or down-regulation of processing or interaction with LIR-containing proteins. For instance, it was shown that loss of phosphorylation of LC3B at the residue Thr (50) blocks autophagy by impairing the formation of autophagosomes35. On the other hand, phosphorylation of serine/threonine 12 by protein kinase A (PKA) contributes for the loss of LC3 activity36. These results suggest that dynamic phosphorylation is a key step in controlling LC3 activity thus offering the opportunity for therapeutic targeting.
A very recent study has also shown that de-acetylation of LC3 at lysine residues K49 and K51 by SIRT1 mediates shuttling of LC3 from the nucleus to the cytoplasm under starvation conditions. This shuttling is essential for conjugation to PE and incorporation into the autophagic membranes37. Therefore, it might be possible to target the acetylation/de-acetylation reaction and/or the nucleo-cytoplasmic shuttling of LC3.
Overall, the identification of post-translational modifications of LC3 and other autophagy proteins is an intense area of research and it can be expected that further studies will shed more light into the relevance of such modifications in health and disease.
Conclusions
In summary, the processing of LC3 and interactions of LC3 isoforms with the autophagy core machinery and adapter proteins may be very vulnerable to drug targeting. As this constitutes the core process of autophagosome formation, we propose that targeting LC3 and its interactions represents the Achilles’ Heel that ultimately enables the development of effective autophagy-modulating chemicals and biologics.
Acknowledgements: This work was supported by the Medical Research Council UK and BBSRC (to RK).
Biographies
 JOANA R. COSTA is a post-doc at the Translational Research Resource Centre (TRRC) at University College London. She graduated in Molecular and Cell Biology at the Faculty of Sciences and Technology (University Nova de Lisboa) in Portugal and recently, she completed her PhD studies in Molecular and Cell Biology in a collaboration between Imperial College London and the Faculty of Medical Sciences in Lisbon. Since then, she has been working with Dr. Robin Ketteler in the TRRC where she performs high-throughput screening projects.
JOANA R. COSTA is a post-doc at the Translational Research Resource Centre (TRRC) at University College London. She graduated in Molecular and Cell Biology at the Faculty of Sciences and Technology (University Nova de Lisboa) in Portugal and recently, she completed her PhD studies in Molecular and Cell Biology in a collaboration between Imperial College London and the Faculty of Medical Sciences in Lisbon. Since then, she has been working with Dr. Robin Ketteler in the TRRC where she performs high-throughput screening projects.
 JACOB (JACK) HEINTZE is a final year PhD student in the Ketteler group. He obtained his undergraduate in biochemistry (International) at the University of Leeds and spent one Erasmus year in Valencia (Spain). For his PhD project, he studies links between metabolic pathways and the regulation of autophagy. Jack is passionate about science communication and the potential of 3D printing technology in advancing many aspects of science.
JACOB (JACK) HEINTZE is a final year PhD student in the Ketteler group. He obtained his undergraduate in biochemistry (International) at the University of Leeds and spent one Erasmus year in Valencia (Spain). For his PhD project, he studies links between metabolic pathways and the regulation of autophagy. Jack is passionate about science communication and the potential of 3D printing technology in advancing many aspects of science.
 ROBIN KETTELER is group leader and manager of the Translational Research Resource Centre (TRRC) at University College London. The facility uses functional genomics and high-throughput/high-content screening methods to gain insight into cell biology processes such as cell signaling and autophagy. He studied biochemistry at the Free University Berlin and obtained his PhD from the Max-Planck-Institute for Immunobiology in Freiburg, Germany. After a Postdoc at the Massachusetts General Hospital in Boston, he established the TRRC in 2009 in London.
ROBIN KETTELER is group leader and manager of the Translational Research Resource Centre (TRRC) at University College London. The facility uses functional genomics and high-throughput/high-content screening methods to gain insight into cell biology processes such as cell signaling and autophagy. He studied biochemistry at the Free University Berlin and obtained his PhD from the Max-Planck-Institute for Immunobiology in Freiburg, Germany. After a Postdoc at the Massachusetts General Hospital in Boston, he established the TRRC in 2009 in London.
References
- Choi, A. M.; Ryter, S. W.; Levine, B. The New England journal of medicine 2013, 368, 1845-6.
- Laddha, S. V.; Ganesan, S.; Chan, C. S.; White, E. Molecular cancer research : MCR 2014, 12, 485-90.
- Yue, Z.; Jin, S.; Yang, C.; Levine, A. J.; Heintz, N. Proceedings of the National Academy of Sciences of the United States of America 2003, 100, 15077-82; Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E. L.; Mizushima, N.; Ohsumi, Y.; Cattoretti, G.; Levine, B. The Journal of clinical investigation 2003, 112, 1809-20.
- Lassen, K. G.; Kuballa, P.; Conway, K. L.; Patel, K. K.; Becker, C. E.; Peloquin, J. M.; Villablanca, E. J.; Norman, J. M.; Liu, T. C.; Heath, R. J.; Becker, M. L.; Fagbami, L.; Horn, H.; Mercer, J.; Yilmaz, O. H.; Jaffe, J. D.; Shamji, A. F.; Bhan, A. K.; Carr, S. A.; Daly, M. J.; Virgin, H. W.; Schreiber, S. L.; Stappenbeck, T. S.; Xavier, R. J. Proceedings of the National Academy of Sciences of the United States of America 2014, 111, 7741-6.
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; Ryujin, F.; Yoshioka, S.; Nishiyama, K.; Kondo, Y.; Tsurusaki, Y.; Nakashima, M.; Miyake, N.; Arakawa, H.; Kato, M.; Mizushima, N.; Matsumoto, N. Nature genetics 2013, 45, 445-9, 449e1.
- Cullup, T.; Kho, A. L.; Dionisi-Vici, C.; Brandmeier, B.; Smith, F.; Urry, Z.; Simpson, M. A.; Yau, S.; Bertini, E.; McClelland, V.; Al-Owain, M.; Koelker, S.; Koerner, C.; Hoffmann, G. F.; Wijburg, F. A.; ten Hoedt, A. E.; Rogers, R. C.; Manchester, D.; Miyata, R.; Hayashi, M.; Said, E.; Soler, D.; Kroisel, P. M.; Windpassinger, C.; Filloux, F. M.; Al-Kaabi, S.; Hertecant, J.; Del Campo, M.; Buk, S.; Bodi, I.; Goebel, H. H.; Sewry, C. A.; Abbs, S.; Mohammed, S.; Josifova, D.; Gautel, M.; Jungbluth, H. Nature genetics 2013, 45, 83-7.
- Lazarus, M. B.; Novotny, C. J.; Shokat, K. M. ACS chemical biology 2015, 10, 257-61.
- Dowdle, W. E.; Nyfeler, B.; Nagel, J.; Elling, R. A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; Cantwell, J.; Luu, C.; Cornella-Taracido, I.; Harrington, E.; Fekkes, P.; Lei, H.; Fang, Q.; Digan, M. E.; Burdick, D.; Powers, A. F.; Helliwell, S. B.; D’Aquin, S.; Bastien, J.; Wang, H.; Wiederschain, D.; Kuerth, J.; Bergman, P.; Schwalb, D.; Thomas, J.; Ugwonali, S.; Harbinski, F.; Tallarico, J.; Wilson, C. J.; Myer, V. E.; Porter, J. A.; Bussiere, D. E.; Finan, P. M.; Labow, M. A.; Mao, X.; Hamann, L. G.; Manning, B. D.; Valdez, R. A.; Nicholson, T.; Schirle, M.; Knapp, M. S.; Keaney, E. P.; Murphy, L. O. Nature cell biology 2014, 16, 1069-79.
- White, E.; DiPaola, R. S. Clinical cancer research : an official journal of the American Association for Cancer Research 2009, 15, 5308-16.
- Kimmelman, A. C. Genes & development 2011, 25, 1999-2010.
- Frake, R. A.; Ricketts, T.; Menzies, F. M.; Rubinsztein, D. C. The Journal of clinical investigation 2015, 125, 65-74.
- Thorburn, A. PLoS biology 2014, 12, e1001967.
- He, H.; Dang, Y.; Dai, F.; Guo, Z.; Wu, J.; She, X.; Pei, Y.; Chen, Y.; Ling, W.; Wu, C.; Zhao, S.; Liu, J. O.; Yu, L. The Journal of biological chemistry 2003, 278, 29278-87.
- Marino, G.; Fernandez, A. F.; Cabrera, S.; Lundberg, Y. W.; Cabanillas, R.; Rodriguez, F.; Salvador-Montoliu, N.; Vega, J. A.; Germana, A.; Fueyo, A.; Freije, J. M.; Lopez-Otin, C. The Journal of clinical investigation 2010, 120, 2331-44.
- Ketteler, R.; Seed, B. Autophagy 2008, 4, 801-6.
- Pankiv, S.; Clausen, T. H.; Lamark, T.; Brech, A.; Bruun, J. A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. The Journal of biological chemistry 2007, 282, 24131-45.
- von Muhlinen, N.; Thurston, T.; Ryzhakov, G.; Bloor, S.; Randow, F. Autophagy 2010, 6, 288-9.
- Kirkin, V.; Lamark, T.; Sou, Y. S.; Bjorkoy, G.; Nunn, J. L.; Bruun, J. A.; Shvets, E.; McEwan, D. G.; Clausen, T. H.; Wild, P.; Bilusic, I.; Theurillat, J. P.; Overvatn, A.; Ishii, T.; Elazar, Z.; Komatsu, M.; Dikic, I.; Johansen, T. Molecular cell 2009, 33, 505-16.
- Wild, P.; Farhan, H.; McEwan, D. G.; Wagner, S.; Rogov, V. V.; Brady, N. R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; Dotsch, V.; Bumann, D.; Dikic, I. Science 2011, 333, 228-33.
- von Muhlinen, N.; Akutsu, M.; Ravenhill, B. J.; Foeglein, A.; Bloor, S.; Rutherford, T. J.; Freund, S. M.; Komander, D.; Randow, F. Molecular cell 2012, 48, 329-42.
- Lee, H. S.; Daniels, B. H.; Salas, E.; Bollen, A. W.; Debnath, J.; Margeta, M. PloS one 2012, 7, e36221; Daniels, B. H.; McComb, R. D.; Mobley, B. C.; Gultekin, S. H.; Lee, H. S.; Margeta, M. The American journal of surgical pathology 2013, 37, 1014-21.
- Chen, S.; Jiang, Y. Z.; Huang, L.; Zhou, R. J.; Yu, K. D.; Liu, Y.; Shao, Z. M. Clinical cancer research : an official journal of the American Association for Cancer Research 2013, 19, 6853-62.
- Yang, S.; Kimmelman, A. C. Autophagy 2011, 7, 912-3.
- Yoshioka, A.; Miyata, H.; Doki, Y.; Yamasaki, M.; Sohma, I.; Gotoh, K.; Takiguchi, S.; Fujiwara, Y.; Uchiyama, Y.; Monden, M. International journal of oncology 2008, 33, 461-8.
- Wang, J.; Pan, X. L.; Ding, L. J.; Liu, D. Y.; Da-Peng, L.; Jin, T. PloS one 2013, 8, e69038.
- Winardi, D.; Tsai, H. P.; Chai, C. Y.; Chung, C. L.; Loh, J. K.; Chen, Y. H.; Hsieh, C. L. BioMed research international 2014, 2014, 723176; Zhao, H.; Yang, M.; Zhao, J.; Wang, J.; Zhang, Y.; Zhang, Q. Medical oncology 2013, 30, 475.
- Othman, E. Q.; Kaur, G.; Mutee, A. F.; Muhammad, T. S.; Tan, M. L. Journal of clinical laboratory analysis 2009, 23, 249-58.
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. Molecular biology of the cell 2008, 19, 4651-9.
- Fernandez, A. F.; Lopez-Otin, C. The Journal of clinical investigation 2015, 125, 33-41.
- Nguyen, T. G.; Honson, N. S.; Arns, S.; Davis, T. L.; Dhe-Paganon, S.; Kovacic, S.; Kumar, N. S.; Pfeifer, T. A.; Young, R. N. Assay and drug development technologies 2014, 12, 176-89; Shu, C. W.; Madiraju, C.; Zhai, D.; Welsh, K.; Diaz, P.; Sergienko, E.; Sano, R.; Reed, J. C. Journal of biomolecular screening 2011, 16, 174-82; Ketteler, R.; Sun, Z.; Kovacs, K. F.; He, W. W.; Seed, B. Genome biology 2008, 9, R64.
- Sandilands, E.; Serrels, B.; Wilkinson, S.; Frame, M. C. EMBO reports 2012, 13, 733-40.
- Nakatogawa, H.; Ohbayashi, S.; Sakoh-Nakatogawa, M.; Kakuta, S.; Suzuki, S. W.; Kirisako, H.; Kondo-Kakuta, C.; Noda, N. N.; Yamamoto, H.; Ohsumi, Y. The Journal of biological chemistry 2012, 287, 28503-7.
- Birgisdottir, A. B.; Lamark, T.; Johansen, T. Journal of cell science 2013, 126, 3237-47.
- Liu, C.; Ma, H.; Wu, J.; Huang, Q.; Liu, J. O.; Yu, L. BMC cell biology 2013, 14, 27.
- Wilkinson, D. S.; Jariwala, J. S.; Anderson, E.; Mitra, K.; Meisenhelder, J.; Chang, J. T.; Ideker, T.; Hunter, T.; Nizet, V.; Dillin, A.; Hansen, M. Molecular cell 2015, 57, 55-68.
- Cherra, S. J., 3rd; Kulich, S. M.; Uechi, G.; Balasubramani, M.; Mountzouris, J.; Day, B. W.; Chu, C. T. The Journal of cell biology 2010, 190, 533-9.
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; Liu, W. Molecular cell 2015, 57, 456-66.
- Rogov, V. V.; Suzuki, H.; Fiskin, E.; Wild, P.; Kniss, A.; Rozenknop, A.; Kato, R.; Kawasaki, M.; McEwan, D. G.; Lohr, F.; Guntert, P.; Dikic, I.; Wakatsuki, S.; Dotsch, V. The Biochemical journal 2013, 454, 459-66.
Related topics
Disease research, Neurosciences, Oncology, Protein, Therapeutics