

A pragmatic approach to hit validation following biochemical high-throughput screening
Posted: 12 December 2017 | Dr Agnes Martin, Dr Laurent Rigoreau, Dr Sheila McLoughlin | No comments yet
Progressing actives from a high-throughput screen (HTS) of a target protein into suitable starting points for lead identification is fraught with pitfalls. The volume and quality of the HTS output is a function of the quality of the screening library, as well as specific properties of both the target and the screening assay…

Typically, following biochemical screening of our in-house library of over 200,000 compounds, around 1,000 actives are whittled down to two or three series, each consisting of a handful of compounds at most, thus resulting in the removal of over 90% of actives.
The cascade of assays (Figure 1) required to validate hits must be tailored to both the target and the screening assays, and requires close collaboration between biologists and medicinal chemists. Here we review the pragmatic approaches we have elaborated as a small drug discovery organisation to validate the output of over 40 biochemical HTS campaigns against cancer targets, and provide a cautionary tale of the hurdles we have encountered along the way.
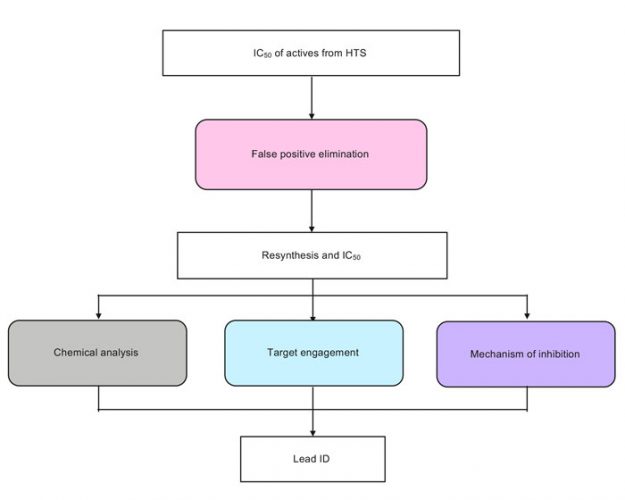
Figure 1: Key steps of a hit validation cascade
Immediately following an HTS, the focus is given to identifying false positives – those compounds that do not achieve inhibition by binding to the target, but employ an alternative non-specific mechanism, and must be eliminated early to prevent wasted efforts. HTS libraries inevitably contain pan-assay interference compounds, or PAINS.1 It is not practical to cherry-pick problematic compounds out of large compound collections; thus, it is preferable to triage out artefacts early and to flag them to facilitate their identification in future screens.
Drug Target Review has just announced the launch of its NEW and EXCLUSIVE report examining the evolution of AI and informatics in drug discovery and development.
In this 63 page in-depth report, experts and researchers explore the key benefits of AI and informatics processes, reveal where the challenges lie for the implementation of AI and how they see the use of these technologies streamlining workflows in the future.
Also featured are exclusive interviews with leading scientists from AstraZeneca, Auransa, PolarisQB and Chalmers University of Technology.
Compounds which interfere with the detection technology can lead to false positives. These can sometimes be identified directly from the primary screening data, for example with a fluorescence ratiometric readout, by examining the raw data in individual channels. Interference assays can also be designed to establish whether test compounds are interfering with the detection technology.
This can either be achieved by spiking the product of the enzymatic reaction and incubating it with compound and detection reagents, or by adding the compound and detection reagents after the enzymatic reaction has been stopped. In addition, orthogonal assays, which employ a different readout to the primary screening assay, are a widely used tool to identify false positives.
Compounds which inhibit the enzyme non-specifically are another source of false positives. Aggregators, for example, may be identified by altering the concentration and type of non-ionic detergent used in the assay.2 Moreover, high Hill coefficients may indicate non-specific inhibition, particularly if several members of a series of compounds display a similar trend.3 In addition, under optimised assay conditions, compound IC50s are expected to be independent of enzyme concentration. As a result, a ratio test can be performed, in which a shift in the IC50 determined at two different enzyme concentrations indicates non-specific binding.4 The effect of enzyme concentration was successfully used in combination with Hill coefficient analysis for compounds screened against PFKFB3 (Figure 2A).5 Whereas the orthogonal assay had not highlighted any concerns, simultaneous analysis of the Hill coefficient and the ratio test allowed the identification of one series in particular, which stood out as having desirable characteristics in both dimensions.
Certain compounds generate reactive products, such as hydrogen peroxide, by redox cycling in the presence of the reducing agents DTT and TCEP, which can lead to oxidation of key residues in the active site of the enzyme and result in false positives. Target classes which are particularly susceptible to this phenomenon include protein tyrosine phosphatases, cysteine proteases, and metalloenzymes. Redox cycling compounds can be identified using an assay which uses horseradish peroxidase to catalyse the oxidation of phenol red by hydrogen peroxide.6 In this way, libraries can be annotated for prompt identification and exclusion of these compounds. However, caution should be exercised, as a false positive in one assay could be a genuine hit in another.
Chemical analysis
Mining historical high-throughput data allows the efficient identification and removal of frequent hitters – promiscuous compounds which are identified as active across multiple high-throughput screens. In addition, the output of the HTS can be mapped in chemical space by clustering through common substructures (Figure 2B). Clusters of compounds increase confidence in an active compound and may allow early structure-activity relationships (SAR) to be established. As a result, clusters would generally be prioritised over singletons. Compounds can then be prioritised based on potency, number of representatives in a cluster, and characteristics of the concentration-response curve. In some instances, contaminants from chemical purification or synthesis have been shown to cause inhibition. For example, residue from SCX columns, routinely used to purify compounds, has been shown to cause false positives under some assay conditions.7 Resynthesis, alternative purification methods, and full analytical characterisation by nuclear magnetic resonance (NMR) and liquid chromatography-mass spectrometry (LC-MS), are key to identify and remove these artefacts, as well as confirm the assigned chemical structure and evaluate the purity of the hits. The low number hit clusters and singletons can be expanded through the purchase of analogues by catalogue (ABCs) to establish SAR.
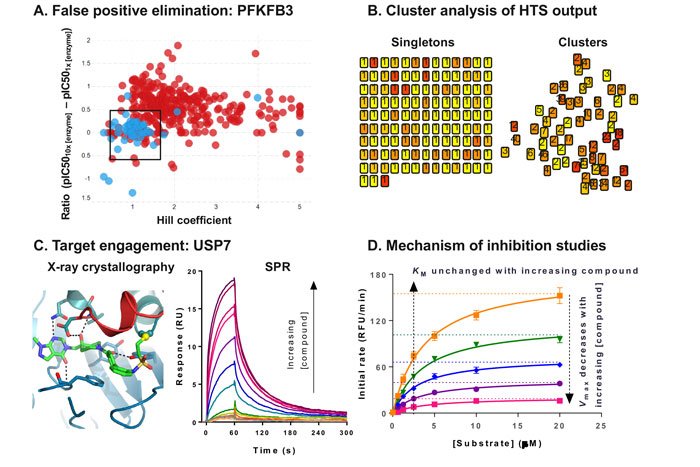
Figure 2: Hit validation data examples
Demonstrating target engagement
Multiple biophysical techniques are available to demonstrate target engagement.8 These have different readouts, as well as different strengths and limitations, such that combinations of assays are used to characterise hits.
X-ray crystallography is the gold standard method to demonstrate target engagement, as it is sensitive and reveals the detailed binding mode. We have recently published the crystal structure of USP7 bound to a novel inhibitor (Figure 2C), and this information was key to progressing this drug discovery programme.9 However, this method does not provide any affinity information, nor is it universally applicable, as some target classes are particularly challenging to crystallise. Moreover, the throughput is low, and increasing automation is an ongoing effort to address this bottleneck.10 As an alternative, NMR can be used to solve structures of protein-ligand complexes for small targets (<40kDa), and this technique can also be used to evaluate compound binding.
Typically, a cascade of assays is used to triage compounds prior to crystallography. For targets that can be immobilised on a chip, surface plasmon resonance (SPR) provides affinity and kinetic measurements, and as it is 384-well plate-compatible, this method is well suited to triaging large numbers of compounds. Differential scanning fluorimetry (DSF) is a high-throughput method detecting thermal stabilisation of a protein target as a result of ligand binding; this cruder method is ideally suited to qualitative triaging. This biophysical technique has also been adapted to enable the detection of target engagement in intact cells, as a cellular thermal shift assay (CETSA).11
Several lower throughput techniques are better suited to characterising small numbers of compounds. Isothermal calorimetry (ITC) is the gold standard label-free method for affinity determination. Although more recent instruments incorporate robotics to increase throughput, the applicability of this technique is often limited by its high protein requirement. Microscale thermophoresis (MST) is a recent technique with applications for hit validation. While the label-free version relies on intrinsic tryptophan fluorescence and is protein-hungry, protein requirements can be decreased significantly when the target protein is fluorescently labelled. Hydrogen-deuterium exchange mass spectrometry (HDX-MS) is a powerful technique to identify the binding site of compound on a protein target, and is particularly useful when x-ray crystallography is challenging.12
Elucidating the mechanism of inhibition
Resolving the mode of inhibition of a compound contributes to building confidence in a hit.4 This can be achieved in a crude, high-throughput manner, for example, by testing whether a compound competes with the substrate by measuring the IC50 at two different substrate concentrations. More detailed characterisation can be performed, albeit in much lower throughput, by analysing the effect of the compound on the affinity of the enzyme for its substrate (Km) and the reaction rate (Vmax) (Figure 2D). From these combined data, it is possible to determine the mode of inhibition of the compound, and whether it preferentially binds to the free enzyme or the enzyme-substrate complex. This information is key to ensure that the relevant form of the enzyme is tested in other assays, from x-ray crystallography to pharmacodynamic biomarker quantification.
While optimisation of affinity is often used as a proxy for in vivo efficacy, the lifetime of the drug-target interaction is a more relevant metric,13 and binding kinetics are commonly measured biophysically, for example, using SPR to directly measure association and dissociation rates of a compound to its target. While often more complicated to design, biochemical methods can also be used to assess binding kinetics, for example kinetic Förster resonance energy transfer (FRET) has recently been reported to measure, in a plate-based format, compound binding kinetics in the context of protein-protein interactions.14
Historically, drug development efforts have shied away from covalent approaches due to safety concerns, although their advantages in terms of prolonged residence time are increasingly recognised.15 Reversibility of inhibition can be tested by saturating the inhibitor binding sites on the enzyme, then measuring the rate of recovery of enzymatic activity following a large, rapid dilution to bring the inhibitor concentration below its IC50. Covalent and slow-binding inhibitors can be crudely identified by measuring the IC50 after two different pre-incubation times. Covalent inhibitors can be characterised biochemically by fitting the non-linear progress curves in the presence of increasing concentrations of inhibitor to extract the affinity of binding and the rate of covalent bond formation. These assays are useful to triage compounds prior to follow up with intact mass spectrometry, peptide fingerprinting, or x-ray crystallography.
Outlook
As the requirement for novel drugs is driving the industry towards difficult targets, developing assays for hit validation is increasingly challenging, and combinations of different assays are essential to increase confidence in hit matter. Moreover, poor validation of tool compounds prior to publication has been cited as a contributing factor to the lack of reproducibility in the field,16 exacerbating project attrition rates. Technological progress is continually increasing the throughput of all steps in the hit validation process, enabling greater numbers of compounds to be triaged, and given the spiralling costs and timescales of drug development,17 stringent validation of hits has become all the more critical to prevent wasting time and money chasing false positives.
Biographies
DR AGNES MARTIN is a Principal Scientist in the Screening and Compound Profiling group at Cancer Research UK’s Therapeutics Discovery Laboratory (CRUK-TDL), specialised in biochemical and biophysical assay development for hit identification and validation. Prior to joining CRUK-TDL in 2013, she worked at Astex Pharmaceuticals, developing biochemical assays to support fragmentbased drug discovery. Agnes completed her PhD in the Department of Pharmacology at the University of Cambridge and her BSc in Biotechnology at Imperial College London.
DR LAURENT RIGOREAU is a Group Leader in the Discovery Chemistry group at CRUK-TDL, specialised in target sourcing and prosecution. He is a veteran of over 25 screening campaigns and leads the development of small molecule projects through hit identification and validation into lead optimisation. Prior to joining CRUK-TDL in 2004, he led the medicinal chemistry on several DNA damage response targets at Kudos Pharmaceuticals.
DR SHEILA McLOUGHLIN is Senior Group Leader for the Screening and Compound Profiling group at CRUK-TDL and project lead for several projects in hit identification. Prior to joining CRUK-TDL in 2008, she spent 20 years developing her expertise in drug discovery at UCB in pharma and biotech, gaining broad experience across several therapeutic areas and multiple target classes.
References
- Baell J, Walters MA. Chemistry: Chemical con artists foil drug discovery. Nature. 2014;513:481-483. doi:10.1038/513481a.
- Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. High-throughput assays for promiscuous inhibitors. Nat Chem Biol. 2005;1:146-148. doi:10.1038/nchembio718.
- Shoichet BK. Interpreting steep dose-response curves in early inhibitor discovery. J Med Chem. 2006;49:7274-7277, doi:10.1021/jm061103g.
- Copeland RA. Evaluation of enzyme inhibitors in drug discovery. 2006. John Wiley & Sons.
- St-Gallay SA, et al. A High-Throughput Screening Triage Workflow to Authenticate a Novel Series of PFKFB3 Inhibitors. SLAS Discov. 2017. Sep 1:2472555217732289. doi:10.1177/2472555217732289.
- Johnston PA, et al. Development of a 384-well colorimetric assay to quantify hydrogen peroxide generated by the redox cycling of compounds in the presence of reducing agents. Assay Drug Dev Technol. 2008;6:505-518. doi:10.1089/adt.2008.151.
- Fillmore MBZ, Brady P, Berlanga-De Lorenzo M, Locke K, Colmenarejo-Sanchez G, Washio Y, Woolven J. In 18th SCI/RSC Medicinal Chemistry Symposium.
- Renaud JP, et al. Biophysics in drug discovery: impact, challenges and opportunities. Nat Rev Drug Discov. 2016;15:679-698. doi:10.1038/nrd.2016.123.
- Turnbull AP, et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550:481-486, doi:10.1038/nature24451.
- Schiebel J, et al. High-Throughput Crystallography: Reliable and Efficient Identification of Fragment Hits. Structure. 2016;24:1398-1409, doi:10.1016/j.str.2016.06.010.
- Martinez Molina D, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84-87. doi:10.1126/science.1233606.
- Campobasso N, Huddler D. Hydrogen deuterium mass spectrometry in drug discovery. Bioorg Med Chem Lett. 2015;25:3771-3776, doi:10.1016/j.bmcl.2015.07.007.
- Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730-739. doi:10.1038/nrd2082.
- Redhead M. in 4th NovAliX Conference: Biophysics in Drug Discovery.
- Bauer RA. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov Today. 2015;20:1061-1073, doi:10.1016/j.drudis.2005.05.005.
- Baker M. Reproducibility: Check your chemistry. Nature. 2017;548:485-488. Doi:10.1038/548485a.
- Workman P, Draetta GF, Schellens JH. Bernards R. How Much Longer Will We Put Up With $100,000 Cancer Drugs? Cell. 2017;168:579-583. Doi:10.1016/j.cell.2017.01.034.
Related topics
Label-free
Related organisations
Cancer Research, Cancer Research UK Therapeutic Discovery Laboratories (CRUK-TDL)
Related people
Dr Anges Martin, Dr Laurent Rigoreau, Dr Sheila McLoughlin