Clinical optimism for antagonists targeting certain ion channels
Adenosine triphosphate (ATP)-gated ion channels serve important neurophysiological functions and have been an attractive class of drug target for a decade…
Of the seven subtypes of ion channels identified to date, active drug discovery programmes have been reported for P2X3, P2X2/3, P2X4 and P2X7 channels. After the early failures of peripherally restricted P2X7 antagonists for inflammatory disorders, there has been a renewed effort to bring forward optimised molecules (clinical candidates) targeting P2X3, P2X4 and P2X7 (with central permeability). This article captures the current clinical landscape of P2X ligands (all antagonists): Afferent’s P2X3 antagonist, AF-219, Nippon Chemiphar’s P2X4 antagonist, NC-2600, and Janssen’s P2X7 antagonist, JNJ-54175446.
The ATP-gated ion channel family (P2X1 – P2X7) has been widely studied in the past two decades, beginning with the skeptical acceptance of ATP as a cellular messenger in the ‘80s to the cloning of mammalian P2X genes, followed by pharmacological and electrophysiological enrichment of the role of these ion channels in (patho)physiology1. In the past 10 years, P2X channels have been heavily prosecuted as drug targets (especially P2X3, P2X2/3, P2X4 and P2X7)2. Of these, P2X7 antagonists from Pfizer and AstraZeneca (peripherally restricted) were the first to advance to the clinic for rheumatoid arthritis (RA) but progression was halted due to lack of efficacy.
While the patent literature was being crowded with novel druggable chemotypes for P2X2/3, P2X4 and P2X7, no new molecules entered into development since the early RA failures, until Afferent Pharmaceuticals (a wholly owned subsidiary of Merck as of July 27, 2016) and Janssen (synonymous with Johnson & Johnson Pharmaceutical R&D, LLC.) pushed P2X3 and P2X7 antagonists (centrally permeable) in the clinic, respectively. In addition, a P2X4 antagonist is reportedly in clinical development as well (Table 1).
P2X ion channels are widely distributed in various tissues; nonetheless, since the endogenous agonist ATP binds to the P2X subtypes with differential affinity and elicits distinct biophysical and electrophysiological properties, all seven P2X ion channels play unique and non-redundant physiological functions. For example, even though P2X7 is ubiquitously expressed, it is mostly a functionally silent channel during normal physiology, as it requires high μM concentrations of ATP (normally absent in the extracellular space) to activate this channel3. Likewise, P2X3 channels are believed to play a significant role in sensitised afferent nerve terminals; a state of the channel that is believed to play a significant role in pathology associated with afferent sensitisation4. Since there have been many comprehensive reviews of P2X ion channels encompassing physiology and pharmacology, the focus of this article is to highlight the recent clinical progress on purinergic ligands, notably antagonists of P2X3 (Afferent/Merck), P2X4 Nippon Chemiphar) and P2X7 (Janssen) (Table 1).
P2X3
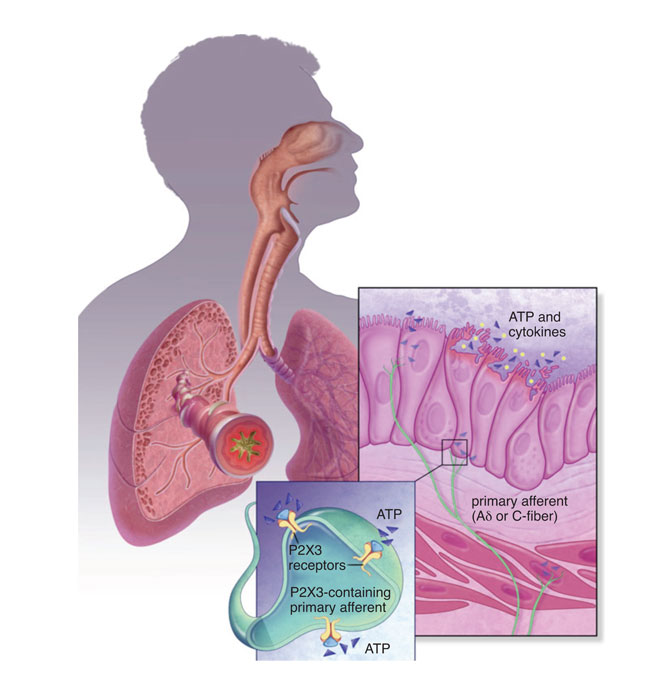
P2X3 channels are expressed predominantly in sensory afferent nerve fibres (C- and Aδ) in most tissues and organ systems, including skin, joints, urinary, bladder and hollow organs of the respiratory system. In addition, P2X3 is expressed pre-synaptically at central terminals of C-fibre afferent neurons. These channels are present either as homotrimers or heterotrimers (P2X2 and P2X3). Preclinical studies from several groups have demonstrated their role in disorders of sensory afferent sensitisation, such as in neuropathic and inflammatory chronic pain, overactive bladder, interstitial cystitis and in respiratory hyper-excitability. A thorough review is outside the scope of this article. Although several companies like Astellas, Merck, AstraZeneca and Evotec have published patents on P2X3 antagonists5, Abbott (now Abbvie) and Roche (now Genentech in the United States) can be credited with the advancement of the P2X3 homomeric and heteromeric (with P2X2) medicinal chemistry leading to the identification and patenting of several drug-like scaffolds.
Abbott was the first to start publishing full research articles, focusing initially on A-3174916, a dual P2X3 & P2X2/3 competitive antagonist. Although a good tool compound, surprisingly, Abbott’s pursuit of it took an unexpected halt, with the relevant publications surrounding its use as a target seeming to disappear. However, the void was filled by the Roche neuroscience group in Palo Alto CA (a site that has now closed due to the Roche-Genentech acquisition), which was led by Dr Anthony Ford (Chief Scientific Officer of Afferent Pharmaceuticals), who disclosed several improved dual antagonists (RO-4, RO-51) with good drug-like properties7,8.
The nomenclature of these molecules became AF-353 and AF-906, after Roche licensed out the series to Afferent Pharmaceuticals. Whether there is a chemical or biological advantage in targeting P2X3 homomeric channels over the heteromers remains to be seen; from all accounts of disclosed press releases and literature, it is believed AF-219, like AF-130, is an antagonist of P2X3 subunit-containing channels. At the time of writing this article, the structure of these antagonists is unknown, although the pharmacology and pharmacokinetics (PK) of AF-219 has been disclosed in a scientific conference (FASEB J 2013; 27 (suppl): 887.5 (abstr)); AF-219 demonstrated some degree of selectivity for P2X3 over P2X2/3 and is believed to be non-competitive with the ATP binding site. Its PK was studied in multiple species and was favourable enough for oral dosing in efficacy models of pain and bladder overactivity. AF-219 demonstrated dose/exposure-dependent efficacy in these models and was pushed through GLP toxicology and CV safety studies for first-in-human dosing (Figure 1).
AF-219 recently demonstrated efficacy in a proof-of-concept study in patients with refractory chronic cough9. This is very exciting news in the field of drug discovery and development; the positive signal is not really ‘surprising’, as the hypothesis of P2X3 antagonism in this affliction has been scientifically backed by clinical and preclinical data. For example, clinical studies have shown that direct ATP application or injection can cause irritation and pain sensations in skin and muscle, and inhalation of ATP can similarly produce airway signs and symptoms, including bronchospasm, coughing, wheezing, dyspnea and chest tightness, especially in patients with respiratory disorders such as asthma or chronic obstructive pulmonary disease.

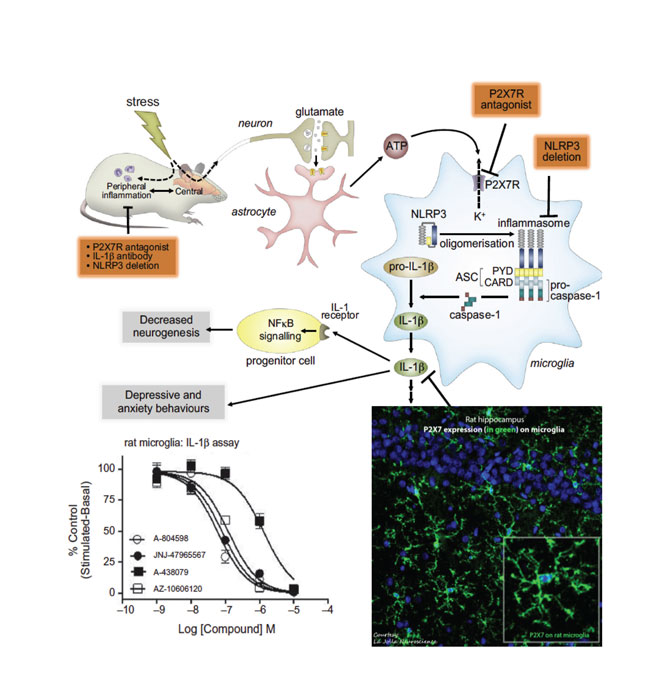
Chronic stress causes mood disorders including depression. ATP-induced activation of P2X7 on channels in CNS microglia during chronic stress in animal models causes release of the proinflammatory cytokine IL-1b. JNJ-54175446, a P2X7 antagonist, is in clinical development for depression. (Adapted from Biol Psychiatry, 2016; 80(1): p. 12-22.)
In preclinical models of cough reflex in guinea pigs, ATP caused sensitisation via P2X3 ion channels. ATP-mediated activation of airway afferents was sensitive to A-317491; this was confirmed by electrophysiology recordings from these neurons as well. In a separate study, AF-353 and TNP-ATP (P2X2/3 and P2X3 antagonists) were effective in attenuating bronchoconstriction induced by C-fibre stimulation in guinea pigs. Hence, there was optimism for a positive clinical cough readout for a P2X3 antagonist, provided the right molecule/dose was tested in the right patient population. And this was indeed the case; in a small sample of patients with chronic cough, AF-219 (600mg; BID for two weeks) produced a significant reduction (75%) in daytime cough frequency. Since the dose and exposure was high in the study, it is not possible to delineate P2X3 over the P2X2/3 component at this time. Lower doses of AF-219 that is P2X2/3-sparing will help to address this.
In addition to chronic cough, AF-219 (50-300mg; BID for four weeks) was also efficacious in reducing urinary urgency in patients with moderate to severe symptoms of interstitial cystitis (www.afferentpharma.com/view.cfm/57/press-release). Nonetheless, there were no serious adverse events in the two-to-four week dosing period in these clinical studies. There were some compound-related taste disturbances, but this should not preclude further plans for clinical development.
In fact, AF-219 continues to surge through the clinical pipeline, with the candidate currently in several trials, including one for idiopathic pulmonary fibrosis (50mg; BID for two weeks). In addition, another P2X3 antagonist (AF-130) is being tested in clinical trials for hypertension, based on undisclosed preclinical data that P2X3 channels on the carotid sinus nerve modulate hypertension in animal species. In summary, AF-219 and AF-130 hold promise in clinical prosecution of P2X3 for disorders of the sensory afferent nervous system.
|
P2X
|
COMPOUND
|
COMPANY
|
DEVELOPMENT
|
INDICATION
|
|
P2X3
|
AF-219/MK-7624
|
Afferent/Merck
|
Phase II
|
Cough, IC, IPF, OA
|
|
P2X3
|
AF-130
|
Afferent/Merck
|
Phase I
|
Hypertension
|
|
P2X4
|
NC-2600
|
Nippon Chemiphar
|
Phase I
|
Neuropathic pain
|
|
P2X7
|
JNJ-54175446
|
Janssen Phase
|
Phase I
|
Mood disorder
|
P2X4
Kyushu University and Nippon Chemiphar Co., Ltd. initiated a Phase 1 clinical trial of NC-2600 in June 2016 (there is no information on structure or preclinical data). The primary indication appears to be neuropathic pain. NC-2600 is a first-in-class candidate designed to control pain by targeting glial cells and is Expected to attenuate neuropathic pain evoked in the peripheral and central nervous system. This molecule was in safety/tolerability studies at the time of writing this article.
P2X4 ion channels are expressed in microglia, the resident immune cells of the central nervous system. In the spinal cord, microglial activation leading to P2X4-dependent neuropathic pain states has been unequivocally established. The pain field has notoriously suffered from lack of bench science-to-bed-side translatability; there are numerous examples, including NK-1 and TRPV1 antagonists. There is no doubt that commercial opportunity for an analgaesic without the horrible side effects of the opiates are huge; yet almost every major pharmaceutical company has left the space due to numerous failed clinical trials and regulatory/reimbursement hurdles. We certainly remain optimistic about NC-2600 and look forward to positive clinical outcomes in the future.
P2X7
As with P2X4, P2X7 is another microglial purinergic ion channel; but unlike P2X4, its activation is linked to the release of pro-inflammatory cytokines IL-1b and IL-18. ATP has the lowest affinity for P2X7 amongst all the purinergic ion channels and, as such, extracellular ATP concentrations seldom activate P2X710. Hence, P2X7 activation occurs ‘on demand’ during cellular stress or insult causing cell necrosis where ATP-mediated activation of P2X7 causes neuroinflammation. An overwhelming body of literature suggests that manipulation of central IL-1b (either by exogenous administration or by selective ablation of signalling by pharmacology or genetics) results in modulation of behaviour, with IL-1b causing animals to behave in a way that resembles clinical depression.
In addition, increased levels of IL-1b have been measured in plasma, cerebrospinal fluid and in the postmortem brain tissue of mood disorder patients. The P2X7 ion channel has recently been directly linked to the occurrence of major depression, with several groups having shown that P2X7 knockout mice demonstrate an antidepressant phenotype. Consistent with knockout animal findings, a number of studies have indicated that antagonism of P2X7 with centrally penetrant antagonists resulted in antidepressant-like behaviour in animal models. In models of chronic stress in animals, it has been shown that increased brain levels of ATP causes anhedonic behaviours in rats through P2X711.
Chronic stress causes mood disorders including depression. ATP-induced activation of P2X7 ion channels in CNS microglia during chronic stress in animal models causes release of the proinflammatory cytokine IL-1b. JNJ-54175446, a P2X7 antagonist, is in clinical development for depression. (Adapted from Biol Psychiatry, 2016; 80(1): p. 12-22.)
Likewise, in another model of depression (BCG model) that has a strong neuroinflammatory component, a P2X7 antagonist reversed the behaviour deficits in mice (Society of Biological Psychiatry 2016 meeting). In addition to depression, patients suffering from bipolar disorder may also benefit from P2X7 antagonism, which may be used to treat the manic phase of that disease. A recent study demonstrated that P2X7 knockout mice showed an attenuated response to amphetamine-induced hyperactivity; a model of manic behaviour. This finding was corroborated with small-molecule antagonists of P2X712. As such, Janssen has progressed a centrally penetrant P2X7 antagonist that is in clinical development (Table 1) with the goal to test it in patients with mood disorders (source: ThomsonReuters IntegritySM). JNJ-54175446 is a P2X7-selective antagonist with low nM potency at both rodent and human orthologues; the compound is orally bioavailable; has a brain/plasma ratio of ~ 1; exhibits dose linearity; and demonstrates central target engagement in a dose/exposure-dependent manner (ACS Fall 2016 meeting in first disclosures section).
In addition to mood disorders, there is the potential of this mechanism to be transformational in epilepsy, particularly as a disease-modifying therapy13. P2X7 is upregulated at the protein level in postmortem brain tissue (neocortex) from patients with temporal lobe epilepsy. Secondly, there are numerous reports in animal models of epilepsy indicating a role of P2X7 activation. As an example, in a model of systemic kainic acid induced tonic–clonic seizures in rats, P2X7 immunoreactivity was enhanced in the rat hippocampus. In a similar mouse model a P2X7 antagonist significantly reduced seizures. Likewise, seizure severity was also reduced in P2X7 knockout mice. In addition to seizure suppression, it also demonstrated protection against neurodegeneration, microglial activation and IL-1b release in epileptic mice treated with P2X7R antagonists. Taken together these data point to a critical role of P2X7 in the pathogenesis of epilepsy in animal models (Figure 2). It remains to be seen whether these preclinical findings translate into clinical efficacy.
Finally, there has been a real push to identify P2X7 ligands for positron emission tomography (PET). P2X7 PET ligands have two potential clinical utilities: firstly, such ligands can be used to accurately access central P2X7 occupancy of clinical compounds for dose selection; secondly, P2X7 PET ligands have the potential to be used as biomarkers of neuroinflammation as P2X7 is up-regulated during microglial activation and neuroinflammation. To that end, [11C]-GSK1482160 and [11C]-JNJ54173717 have recently been published and show promise14,15.
In summary, there is clearly optimism for the clinical success of compounds targeting P2X3, P2X4 and P2X7 ion channels based on their role in animal models of disease.
AF-219 is the furthest along, and with positive clinical signals in two studies (reduction in cough and urinary urgency), the field is waiting for some/ all of these molecules to become accessible to patients after registration and approval.
ANINDYA BHATTACHARYA, PhD is Scientific Director at Janssen Research & Development, based in San Diego, CA. He is a neuropharmacologist and has been involved with neuroscience drug discovery efforts throughout his career: presently for 11 years at Janssen, and previously for four years at Roche in Palo Alto, CA. He obtained his PhD in pharmacology from State University of New York, Buffalo (USA). During his career in the industry, Anindya has worked on several ion channel and GPCR drug targets.
References
- Habermacher C, et al. Molecular structure and function of P2X receptors. Neuropharmacology. 2016; 104: p. 18-30
- Jacobson KA, CE Muller. Medicinal chemistry of adenosine, P2Y and P2X receptors. Neuropharmacology. 2016; 104: p. 31-49
- Bhattacharya A, K Biber. The microglial ATP-gated ion channel P2X7 as a CNS drug target. Glia, 2016; 64(10): p. 1772-87
- Ford AP. In pursuit of P2X3 antagonists: novel therapeutics for chronic pain and afferent sensitization. Purinergic Signal. 2012; 8(Suppl 1): p. 3-26
- Bolcskei H, B Farkas. P2X3 and P2X2/3 receptor antagonists. Pharm Pat Anal, 2014; 3(): p. 53-64
- Jarvis MF, et al. A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc Natl Acad Sci U S A. 2002; 99(26): p. 17179-84
- Carter DS, et al. Identification and SAR of novel diaminopyrimidines. Part 1: The discovery of RO-4, a dual P2X(3)/P2X(2/3) antagonist for the treatment of pain. Bioorg Med Chem Lett. 2009; 19(6): p. 1628-31
- Jahangir A, et al. Identification and SAR of novel diaminopyrimidines. Part 2: The discovery of RO-51, a potent and selective, dual P2X(3)/P2X(2/3) antagonist for the treatment of pain. Bioorg Med Chem Lett, 2009. 19(6): p. 1632-5
- Abdulqawi R, et al. P2X3 receptor antagonist (AF-219) in refractory chronic cough: a randomised, double-blind, placebo-controlled phase 2 study. Lancet. 2015; 385(9974): p. 1198-205
- Rech JC, et al. The evolution of P2X7 antagonists with a focus on CNS indications. Bioorg Med Chem Lett. 2016; 26(16): p. 3838-45
- Iwata M, et al. Psychological Stress Activates the Inflammasome via Release of Adenosine Triphosphate and Stimulation of the Purinergic Type 2X7 Receptor. Biol Psychiatry. 2016; 80(1): p. 12-22
- Bhattacharya A, et al. Pharmacological characterization of a novel centrally permeable P2X7 receptor antagonist: JNJ-47965567. Br J Pharmacol. 2013; 170(3): p. 624-40
- Jimenez-Pacheco A, et al. Transient P2X7 Receptor Antagonism Produces Lasting Reductions in Spontaneous Seizures and Gliosis in Experimental Temporal Lobe Epilepsy. J Neurosci. 2016; 36(22): p. 5920-32
- Ory D, et al. Preclinical Evaluation of a P2X7 Receptor-Selective Radiotracer: PET Studies in a Rat Model with Local Overexpression of the Human P2X7 Receptor and in Nonhuman Primates. J Nucl Med. 2016; 57(9): p. 1436-41
- Territo PR, et al. Characterization Of [11C]-GSK1482160 For Targeting The P2X7 Receptor As A Biomarker For Neuroinflammation. J Nucl Med. 2016