

Researchers identify a gene linked to neurodegenerative disorders – SLC25A46
Posted: 14 July 2015 | Victoria White
Researchers have discovered and characterised a previously unknown disease gene, SLC25A46, linked to the degeneration of optic and peripheral nerve fibres…
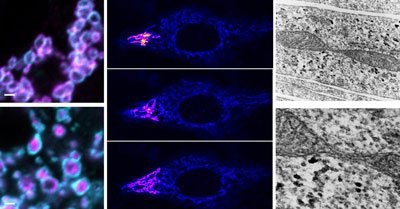
The left panels are high resolution microscope images showing the donut shaped outer mitochondrial membranes. SLC25A46 unlike typical mitochondrial transporters localizes to the outer mitochondrial membrane. Middle panels: Visualization of the inter-connected mitochondrial network by photoactivation of mitochondria in a HeLa cell. Right panels: Electron micrographs showing a mitochondrial constriction site reminiscent of a mitochondrial fission event. CREDIT: Abrams et al 2015
University of Miami (UM) researchers have discovered and characterised a previously unknown disease gene linked to the degeneration of optic and peripheral nerve fibres.
Patients with mutations in this gene present symptoms similar to optic atrophy and Charcot-Marie-Tooth Type 2 (CMT2), including vision loss and weakening of the lower leg and foot muscles beginning in the first decade of life.
The novel variants occur in a gene called SLC25A46 that functions in mitochondria. They transform food into fuel that allow cells to carry out energy-demanding functions.
“Mitochondria play a large role in human health,” said Alexander Abrams, Ph.D. student in Neuroscience at the UM Miller School of Medicine. “Although we study rare diseases such as CMT2 and optic atrophy, the implications encompass all forms of neurodegeneration including Lou Gehrig’s and Parkinson’s Diseases.”
Mitochondria constantly undergo fusion and fission to respond to cellular energy demands. By changing their size and connectivity through fusion and fission, mitochondria can travel to regions in cells where they are needed.
Disrupting SLC25A46 causes mitochondria to become improperly localised in cells
“Our study reveals that disrupting SLC25A46 causes mitochondria to become both more highly interconnected and improperly localised in cells,” said Julia E. Dallman, assistant professor of Biology in the UM College of Arts and Sciences. “These data support a critical role for SLC25A46 and mitochondrial dynamics in the establishment and maintenance of neuronal processes.”
SLC25A46 encodes an atypical protein in the SLC25 family. SLC25 family members act like a channel, transporting molecules across the bilayer membranes inside mitochondria. But unlike the majority of human SLC25 family members (there are 53) that transport molecules across the inner mitochondrial membrane, SLC25A46 settles on the outer mitochondrial membrane where it regulates mitochondrial dynamics.
Mutations in the genes associated with mitochondria dynamics OPA1 and MFN2 are linked to similar mitochondrial disorders. Homologous genes in baker’s yeast, work in combination with a gene called UGO1, which has ancestral similarities to SLC25A46. The new findings suggest that the SLC25A46 and Ugo1 proteins may play similar roles.
Given the similarities between the diseases caused by mutations in OPA1, MFN2 and SLC25A46, these genes could be involved in common pathological mechanisms of neurodegeneration, the study says.
“This finding builds on our discovery of MFN2 as a major disease gene in this area over 10 years ago,” said Dr Stephan Züchner, professor and chair of the Dr John T. Macdonald Foundation Department of Human Genetics, at UM’s Miller School of Medicine. “Only through the new genome sequencing methods and active global data exchange were we able to solve this puzzle.”
The study findings are published in full in Nature Genetics.
Related topics
Gene testing
Related organisations
Cellular Dynamics, Miami University