

Lancet tool enables accurate identification of rare mutations in cancer cells
Posted: 26 March 2018 | Dr Zara Kassam (Drug Target Review) | No comments yet
A new computational method allows scientists to identify rare gene mutations in cancer cells with greater accuracy and sensitivity…
A new computational method developed by researchers allows scientists to identify rare gene mutations in cancer cells with greater accuracy and sensitivity than currently available approaches.
The technique called Lancet and represents a major advance in the identification of tumour cell mutations, a process known as somatic variant calling.
“With its unique ability to jointly analyse the whole genome of a tumour and matched normal cells, Lancet provides a useful tool for researchers to conduct more accurate genome-wide somatic variant calling,” notes first author Dr Giuseppe Narzisi, Senior Bioinformatics Scientist, NYGC.
“Reliable detection of somatic variations is of critical importance in cancer research and increasingly in the clinical setting, where identification of somatic mutations forms the basis for personalised medicine,” said Dr Michael Zody, Senior Director, Computational Biology, NYGC, and senior author of the study. “Lancet will be an important addition to the toolkit of both clinicians and researchers working to advance the field of cancer genomics and improve care for cancer patients.”
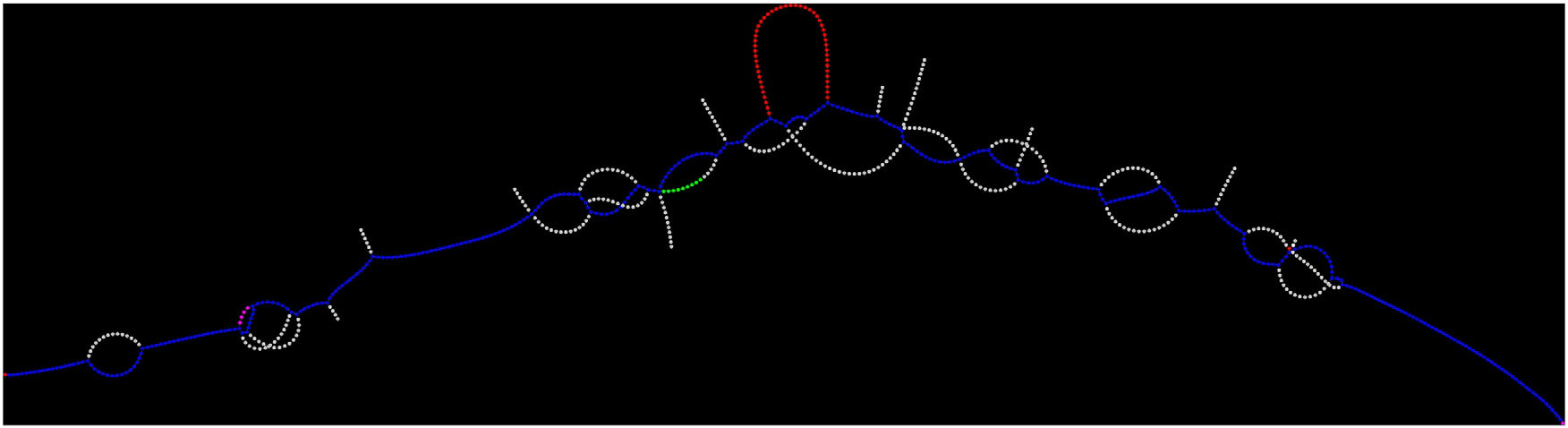
Example of coloured de Bruijn graph rendered to represent genomic data for a short region of 400 base pairs using the Lancet genomics tool developed by New York Genome Center scientists. The somatic variant is highlighted in red. New York Genome Center
To identify gene mutations in cancer cells, researchers sequence the genomes of tumour cells and normal cells. Current computational methods then involve comparing both tumour and normal to a reference genome and looking for differences unique to the tumour. Lancet instead uses an approach called micro-assembly to reconstruct the complete sequences of small regions of the genome without relying on a reference. Because the approach does not rely on a reference to identify variants, it also works well in regions of the genome where comparing reads to a reference is challenging for technical reasons.
By using a data structure called a coloured de Bruijn graph, Lancet jointly analyses the tumour and normal DNA, providing greater sensitivity to find rare variants unique to the tumour while also providing greater accuracy of differentiating tumour variants from those present in healthy tissue in that individual. Using Lancet to combine the sequencing data from the normal and tumour cells represents a more powerful way of identifying mutations, Dr Narzisi said, since users are no longer dependent on analysing sequence data from tumour and normal cells separately.
In the study, through extensive experimental comparison on synthetic and real whole-genome sequencing datasets, the researchers demonstrated that Lancet performed better and had higher accuracy and better sensitivity to detect somatic variants compared to the most widely-used somatic variant callers.
“In our study, we show that existing tools are not that precise in scoring mutations so that some candidate variants which were highly scored by some tools ended up being false positives,” Dr Narzisi said. “That becomes a problem when you want to prioritise which variants to validate using other technologies or you want to move forward with a clinical study. You may end up focusing on variants that do not exist.”
The study has been reported in Communications Biology.
Related topics
Genomics, Imaging, Oncology, Personalised Medicine, Sequencing
Related conditions
Cancer
Related organisations
New York Genome Center (NYGC)
Related people
Dr Giuseppe Narzisi, Dr Michael Zody