

Flow cytometry: Not just a box in the lab
Posted: 21 September 2015 | Rachael Walker (Babraham Institute)
Undoubtedly, you will have noticed the increase in the number of publications utilising flow cytometry, heard it mentioned more in meetings and probably even been targeted by several companies trying to sell you their latest cytometer. The rapidly advancing technology behind the cytometer and an increase in applications that use flow cytometry is what has led to this; the flow cytometry market was reported to be worth $2.3 billion in 2013. It sounds like a lot, but it is forecast to climb still further, to £3.7 billion by 2018.

A powerful technology in biology and medical research, flow cytometry allows for the identification and separation of cells in many fields including pathology, immunology, molecular biology, cell biology, drugscreening, high-throughput assays, stem cell research as well as botany, microbiology, marine biology and veterinary sperm sorting for sex pre-selection. Flow Cytometry is also routinely used for diagnostics in clinical labs, especially in diagnosing haematopoietic diseases such as leukaemia.
Flow cytometry vs. FACS
The first thing to clear up is the use of the term FACS. FACS stands for ‘Fluorescence Activated Cell Sorting’ and is a trade name of Becton Dickinson (BD). FACS is often incorrectly used; it is a bit like saying Biro instead of ball-point pen or Hoover instead of vacuum cleaner. It is correct to call it flow cytometry and these cytometers are separated into analysers and cell sorters. The analysers allow detection of cells at rates of several thousand a second. Cell sorters use the same principles of detection as analysers but the cells can be separated using electrostatic charges or mechanical methods.
The first flow cytometer, a cell sorter, was invented by Mack Fulwyler in Los Alamos in 19652 and incorporated the technology behind ink-jet printing and the Coulter principle3 (detection and counting of cells using impedance through an orifice and an electric field). Since then, cytometers have evolved by the addition of more lasers and the advancement in the technology behind the machine components, antibodies and fluorochromes.
Drug Target Review has just announced the launch of its NEW and EXCLUSIVE report examining the evolution of AI and informatics in drug discovery and development.
In this 63 page in-depth report, experts and researchers explore the key benefits of AI and informatics processes, reveal where the challenges lie for the implementation of AI and how they see the use of these technologies streamlining workflows in the future.
Also featured are exclusive interviews with leading scientists from AstraZeneca, Auransa, PolarisQB and Chalmers University of Technology.
Commercial flow cytometers have been available since the mid-1970s and since then the technology behind the instruments and fluorochromes has advanced tremendously to bring us the sophisticated multi-laser machines we see in labs today. The main companies designing, manufacturing and selling cytometers are BD and Beckman Coulter. However, there are other manufacturers such as Miltenyi Biotec, Sony Biosciences, ACEA Biosciences, Thermo Fisher and Bio-Rad. Each of these machines have unique selling points, different combinations of lasers etc., but they all measure cells (in fact its not just cells that can be measured) using the same principles and looking at the same parameters.
What can we measure on a flow cytometer?
Flow cytometry is a laser based technology which is used to identify and analyse independent events, as well as cell counting, the detection of biomarkers and if required cell separation. It allows multiple parameters on each individual cell to be independently analysed in a heterogeneous population in the absence of other cells, at a rate of several thousand per second. Cytometers can be used to look at a suspension of particles, for example, mammalian cells, organelles such as nuclei, chromosomes or bacteria.
In this article I will concentrate on cells analysed via a flow cytometer. As the name suggests the cells must be in a single cell suspension in a flow cytometer and these cells pass through a path of light of a single wavelength, which is usually a laser. There are a number of detectors aimed at the interrogation point and measurements of the scattering of light and any fluorescence emitted can be made. This allows multiple parameters to be measured simultaneously giving information about each individual particle that passes through the light source.
As each cell passes through the laser beam, the light is scattered as well as fluorescence being detected, if a fluorochromes is present on the cell, which allows the phenotyping of the cell to be identified.
Scatter
Almost all cytometers measure light scattering at small and larger angles to the primary laser beam, which is commonly known as forward scatter and side scatter.
Forward scatter
Forward scatter is scattered light collected at small angles of between 0.5° and 5° to the incident beam. Forward scatter is often attributed to the size of a cell, however this is incorrect. It is not possible to accurately measure the size of a cell on a flow cytometer. Forward Scatter is not an accurate measurement of cell size because the insensitivity of the signal is strongly affected by the wavelength of light used and by the precise range of angles over which light is collected, the refractive index between cells and the suspending medium as well as number of other factors. For more information about light scatter, there are a couple of interesting chapters in Howard Shapiro’s Practical Flow Cytometry book4.
Side scatter
Side scatter sheds light on the internal granular structure of cells. Light is scattered all around and then a particle passes through the laser beam and light that scatters at larger angles of 15° to 150° to incident beam is called side scatter (also known in some books as right-angle scatter or orthogonal scatter). In a majority of cytometers, side scatter is collected at 90° to the laser beam.
The combination of forward and side scatter is used in nearly all experiments to look at heterogeneous cell populations and to be able to distinguish different cell types within. For example, it is possible to distinguish the different population of leukocytes in peripheral blood mononuclear cells (PBMCs) using scatter (Figure 1).
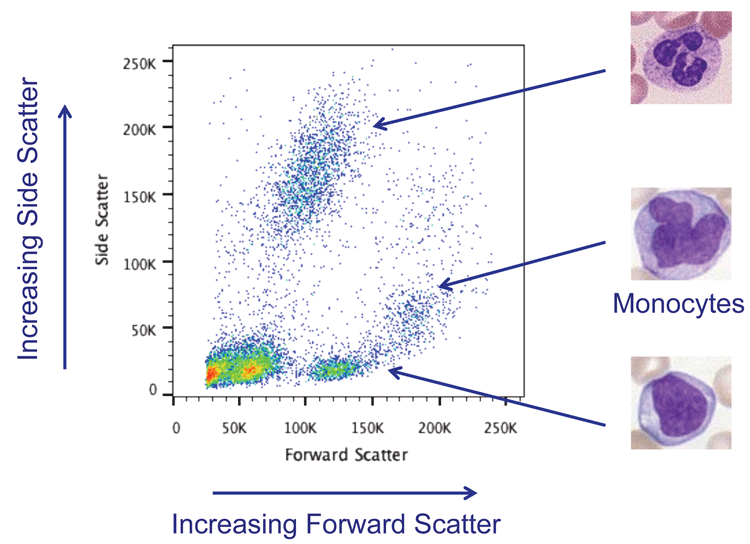
Figure 1: Forward and Side Scatter plot showing lysed whole blood. The different types of white blood cells can be distinguished using scatter.
Fluorescence
Cells have specific functional antigens on their surface and as well as intracellular antigens which can be identified using specific antibodies to those antigens. This allows the cells to be phenotyped. It is akin to having a ‘barcode’ on the cells.
Fluorochromes are molecules, which can be excited by specific wavelengths of light, which cause these molecules to raise their energy levels to a brief excited state. These molecules return to ground state by emitting a photon of light of a longer wavelength than the photon that excited it, which is essentially what fluorescence is. It is possible to use several fluorochromes per laser allowing multiple fluorescence to be detected. By adding fluorochromes to these antibodies it is possible to simultaneously detect multiple antigens on a cell and therefore to detect rare subpopulations of cells and several cell types simultaneously.
Different fluorochromes can be excited by differing wavelengths of light and emit photos of different wavelengths of light and it is using combinations of these fluorochromes that allows scientists to do ‘multi-colour’ experiments. Fluorescent probes can also be used to provide a sensitive method for determining the structure, function and health of cell, especially since it is possible to detect only a few fluorescent molecules per cell.
How a cytometer works
In cytometry, a suspension of single cells is introduced into the cytometer in a tube (which can either be pressured or delivered by a syringe, depending on which cytometer you are using). A majority of cytometers use a sheath fluid (usually PBS) to introduce the cells in the flow cell. The sheath fluid hydrodynamically focuses the sample so that the cells are introduced one by one through the laser beam. This happens in the flow cell of the cytometer.
The flow cell is the most important part of a cytometer, since this is where each and every cell (actually every event) passes through the laser beam(s). If a cytometer has multiple lasers, these can either be co-linear, where the laser beams overlap and go through the flow cell at the same point or are spatially separated. It is important to know how your lasers are arranged since this can have implications in your fluorochrome choice, e.g., if you have a cytometer with a co-linear 488nm blue laser and 635nm red laser then you cannot use Pe-cy5 and APC together in an experiment.
For every cell that goes through the cytometer, information about the scatter and fluorescence is collected. As mentioned earlier forward scatter is collected straight ahead and side scatter and fluorescence are usually collected at 90° to the laser beam.
Any photons of light emitted during fluorescence pass through a series of optical filters, which separate out the different wavelengths of light and steer the photons towards the detectors. In front of the detectors are bandpass filters, which allow specific wavelength of light through them. For example, when detecting GFP (emits at 510nm), a 530/30 bandpass filter is used in front of the detector, which allows light between 515 and 545nm to be collected. Even though it does not quite cover the emission maxima, the signal will be bright enough at 515nm to collect the photos.
There are two main types detector in a cytometer, photodiodes and photomultiplier tubes (PMT). Photodiodes are used to collect photons for bright signals such as forward scatter. PMTs are used to collect the side scatter signal and any photons emitted. PMTs convert photons into an electrical signal.
How your data is generated
For every cell that passes through the cytometer, a voltage pulse is generated in every parameter. That is, after photons strike a PMT, they are converted into an electrical current which then travels to the amplifier and is converted into a voltage pulse. The highest part of the pulse occurs when the cells are in the middle of the laser beam. For each pulse the height, width and area of it is measured. The size of the pulse depends on the number of photons that have been detected and the voltage settings of the PMT. A voltage can be applied to a PMT to amplify a signal as this will create a larger electrical current.
Figure 2 (page 00) depicts the signal from the PMT through pre-amplifiers before it goes through an analog-to-digital converter (ADC). Each pulse for each parameter is given a numerical value and assigned a channel number on a data plot. This value is plotted and these values are proportional to light scatter and fluorescence intensity of the event. The resolution of the data will depend on the ADC, for example an 8-bit converter will give 256 channels and a 10-bit converter gives 1024 channels.
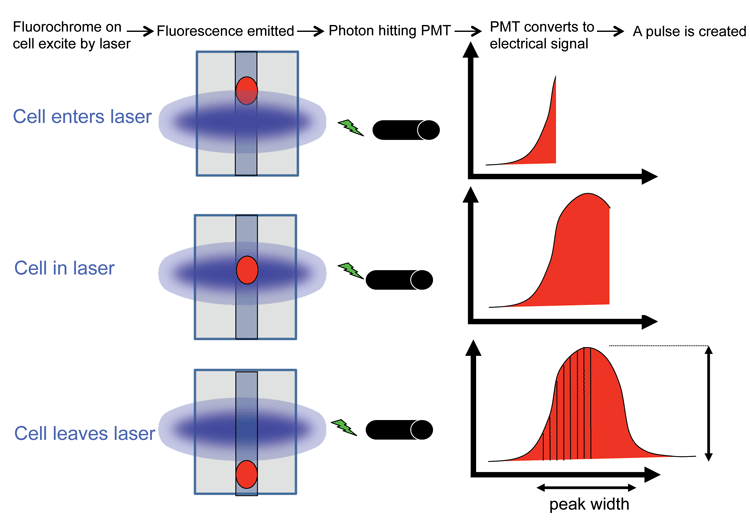
Figure 2: Diagram showing how a pulse is created as a cell passes through the laser beam. For every cell that passes through the laser beam a pulse is created in every parameter being measured. The height, width and area of the pulse is recorded. Credit: Rachael Walker, Babraham Institute.
Data files
For every cell that passes through the cytometer, the values from every parameter are stored as a flow cytometry standard (FCS) files. The FCS format was developed for the International Society for the Advancement of Cytometry (ISAC)5. The data is saved as a list-mode file which means that it lists the parameters and their measurements on a cell-by-cell basis. It is that data that is plotted on histograms or dotplots that you will see presented in lab meetings or in papers. The signals can be measured in a linear or logaritimic scale depending on the application.
In summary, knowing a bit more about the ‘box’ in your lab will allow you to design and carry out better experiments on your cytometer. There are a whole host of applications that can be carried out on a cytometer. So switch on the cytometer and ‘go with the flow’.
References:
- http://www.marketsandmarkets.com/Market-Reports/flow-cytometry-market-65374584.html).
- Fulwyler MJ. Electronic Separation of Biological Cells by Volume. Science. 1965; 150, 3698 910-911
- Coulter WH. High Speed Automatic Blood Cell Counter and Cell Size Analyzer. Proceedings of the National Electronics Conference. 1956; 12:1034-1042, 1957
- Shapiro HM. Practical Flow Cytometry: 4th 2003. Chapter 1 and 7. Wiley-Liss http://www.coulterflow.com/bciflow/practical/book/index.html
- http://isac-net.org/Resources-for-Cytometrists/Data-Standards/Data-File-Standards/Flow-Cytometry-Data-File-Format-Standards.aspx
Biography
Dr Rachael Walker is Head of Flow Cytometry at the Babraham Institute, Cambridge, with over 13 years’ experience with flow cytometry, including over 10 years of running flow cytometry core facilities in several labs in Cambridge.
Dr Walker’s first degree was in Biomedical Materials Science from the University of Birmingham. Her first experience with flow cytometry was during her PhD in Tissue Engineering at the University of Liverpool and she then decided that she wanted to pursue a career in a flow cytometry. After completing her PhD, Rachael worked in the flow cytometry flow facility at the Cambridge Institute for Medical Research, followed by five years managing the Flow Cytometry Facility at the Cambridge University Stem Cell Institute. She joined Babraham Institute in 2012. Rachael is a very active member of the cytometry world and is a member of ISAC, flowcytometryUK and the Royal Microscopical Society. In 2012 she was awarded a five-year ISAC Scholarship/Shared Resource Laboratory Emerging Leader award. Rachael was written and hosts flow cytometry training courses aimed at scientists using flow cytometry in Industry. See: http://www.babraham.ac.uk/science-services/flow-cytometry/flow-training
Related topics
Flow Cytometry, High Throughput Screening (HTS), Immunology, Screening
Related organisations
Babraham Institute
Related people
Rachael Walker