Novel approaches to targeting protein tyrosine kinases
Protein kinases, a diverse group of cellular enzymes, play fundamental roles in maintaining normal cellular functions such as growth, cell cycle control, proliferation, differentiation, migration, cellular survival and apoptotic induction.
They participate in a variety of cellular signalling cascades, such as the Ras/Raf/MAPKinase1-4 and PI3Kinase pathways5-8. After G-protein-coupled receptors, protein kinases are considered the second most important class of drug targets. Abnormal kinase activities have been linked either directly or indirectly to over 400 human diseases, particularly those which display proliferative or inflammatory characteristics. Examples include neurological and cardiovascular diseases, psoriasis, asthma, rheumatoid arthritis and crucially cancer9.
Upon ligand binding, protein kinases alter the properties or functional characteristics of a variety of substrate proteins. By catalysing the transfer of γ-phosphate groups from adenosine triphosphate (ATP), kinases alter the activity of substrate proteins in a process termed phosphorylation. Kinases predominantly phosphorylate threonine, serine or tyrosine residues within a substrate’s amino acid (aa) sequence and so can be broadly sub-categorised as either serinethreonine kinases or tyrosine kinases10. A small proportion of kinases can show dual-specificity, acting to phosphorylate serine, threonine and tyrosine, or alternatively can act on other amino acids such as the histidine kinases.
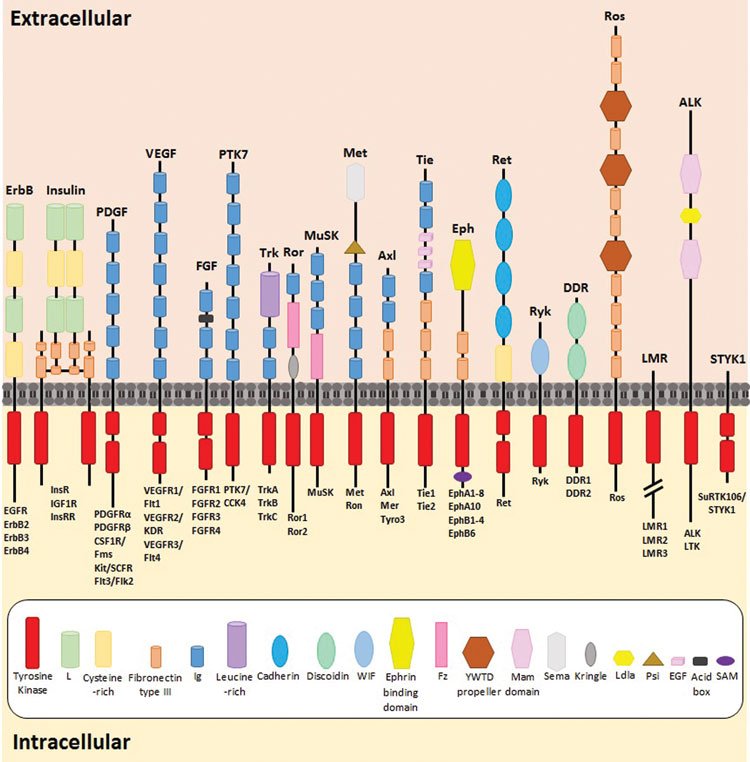
Tyrosine kinases constitute a large proportion of the protein kinases and can be sub-divided into two families, 58 of which are receptor tyrosine kinases (RTKs) and 32 being non-receptor tyrosine kinases (n-RTKs)11. RTKs comprise a highly conserved structure, including a highly glycosylated extracellular domain containing a ligand binding region, a transmembrane domain and a C-terminal intracellular domain composed of a juxtamembrane region, a catalytic tyrosine kinase domain and a carboxyl terminal tail12,13. The structure and composition of the receptor’s extracellular domain dictates its classification into its relevant sub-group, with fibronectin type-III, cysteine-rich motifs and immunoglobulin-like repeats as examples of extracellular domain characteristics. RTK subfamilies include the epidermal growth factor (erbB/HER) receptors, vascular endothelial growth factor receptors (VEGFRs), fibroblast growth factor receptors (FGFR’s), insulin receptors, Met receptors (hepatocyte growth factor/scatter factor [HGF/SF]) and ephrin (Eph) receptors, amongst others11 (Figure 1).

Figure 1: The highly conserved structure of all 58 RTKs, with the individual extracellular characteristic subgroups and overall domain structures. The composition of each RTK’s extracellular domain dictates its classification into its relevant sub-family. Figure adapted from 3
RTKs have been found to be overexpressed or mutated in some of the most prevalently occurring and high mortality tumour types, including breast, lung, colorectal, pancreatic and gastric cancers. Often, up-regulations or mutations in RTKs are indicative of a poorer patient prognosis or a cancer which presents with a more aggressive tumour phenotype. As a result they have become attractive targets for the development of new anti-oncogenic drugs(14). To date, inhibition of RTK activity is predominantly achieved by either targeting the extracellular domain of the receptor or by hindering the catalytic potential of the intracellular kinase. These outcomes can be achieved either by specifically preventing a native ligand, essential substrate or ATP from binding to its relevant domain or by acting as a steric hindrance to receptor interactions.
Small molecule tyrosine kinase inhibitors
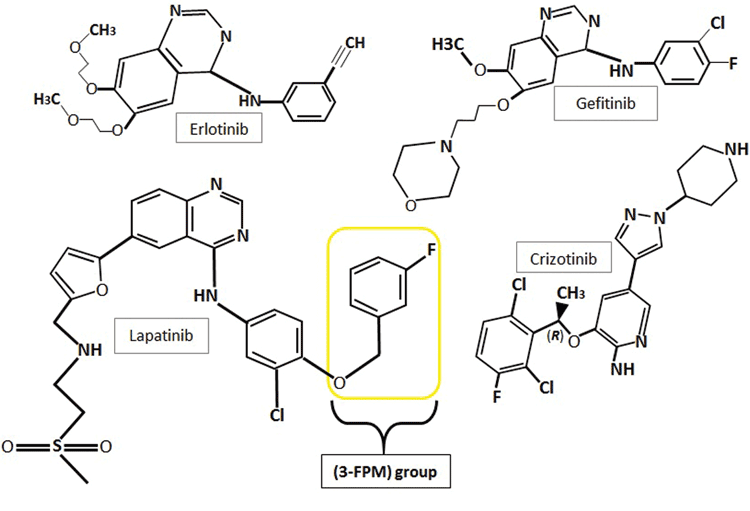
With the discovery of receptor tyrosine kinases came the development of first generation tyrosine kinase inhibitors (TKIs) for the targeted treatment of over-expressing cancer forms. Three first generation examples of TKIs are gefitinib (Iressa®), erlotinib (Tarceva®) and crizotinib (Xalkori®) (Figure 2). These three TKIs are approved for the treatment of non-small cell lung cancers (NSCLC) carrying specific RTK mutations: EGFR in the first two instances and anaplastic lymphoma kinases (ALKs) and c-Met tyrosine kinases in the latter(15). Gefitinib and erlotinib, both quinazoline derivatives, reversibly bind to EGFR’s catalytic tyrosine kinase, forming a complex with its ATP-binding pocket(14). Erlotinib functions as a kinase inhibitor by forming a hydrogen bond between its quinazoline heterocyclic group and the -NH group of amino acid MET793, located within the hinge region of the kinase’s ATP-binding domain(15). Binding of these TKIs to the ATP binding cleft in place of ATP inhibits the receptor’s autophosphorylation and blocks downstream signalling cascades(16).
Figure 2: The chemical structures of the first generation tyrosine kinase inhibitors, erlotinib, gefitinib and crizotinib and second generation inhibitor lapatinib showing their heterocyclic structures and quinazoline rings. Lapatinib also comprises a 3-flourophenylmethoxy (3-FPM) group. Images adapted from 5,16,32
Crizotinib is another TKI approved for the treatment of NSCLCs that present with novel transforming genes. These genes are composed of a fusion between echinoderm microtubule-associated protein-like 4 (EML4) encoding gene fragments and anaplastic lymphoma kinase (ALK) encoding genes. These fusion-gene fragments ultimately result in constitutive kinase activation. Crizotinib comprises an aminopyrimidine structure and acts as a dual small-molecule TKI, binding to the ATP-pockets of two target kinase types, ALKs and c-Met kinases, as well as c-ros oncogene 1 fusion variants (ROS1)(17,18).
Since the development of first-generation TKIs, there has been substantial work to develop more effective second- and third-generation inhibitors. These later generation inhibitors aim to overcome the resistance of their first generation counterparts and ultimately prevent treatment failure. Second-generation TKIs, such as lapatinib (Tyverb®) and afatinib (Giotrif®) have the advantage of blocking multiple RTKs simultaneously.
Lapatinib, approved for the treatment of advanced/metastatic HER2-expressing breast cancers, is an ATP-competitive inhibitor. Lapatinib shows dual-targeted kinase sensitivity, reversibly binding to both ErbB1/EGFR and ErbB2/HER2. Lapatinib treatment is undertaken in patients where a previous treatment has failed(19,20). As with erlotinib, the intracellular activity of lapatinib stems from its capacity to reversibly bind to and block the ATP-binding site of the tyrosine kinase. Also like erlotinib, lapatinib’s inhibition occurs through hydrogen-bond interactions between its quinazoline heterocyclic group and the -NH group of MET793 in the receptor ATP-binding cleft. Simultaneously, lapatinib’s anilino group extends into the hydrophobic region found adjacent to the adenine-binding cleft. The anilino group of lapatinib comprises a 3-flourophenylmethoxy (3-FPM) group (Figure 2), which displaces the αC-helix to an out-position and forces the hydrophobic pocket to extend to the adenine-binding site’s activation segment. Unlike erlotinib which binds to the active conformation of EGFR, lapatinib binds to the displaced inactive αC-out confirmation of EGFR(14,15). Lapatinib is also used for the treatment of breast cancer with secondary brain metastases, due to its ability to effectively cross the blood-brain barrier(21).
Antibody-based RTK therapy
Antibody-based agents have become attractive immunotherapeutic compounds for the targeted treatment of diseases such as cancer. Cancerous cells often display aberrant RTK expression or disease-specific antigens and so a variety of antibodies and antibody-conjugated compounds have been clinically developed to target membrane-bound tyrosine kinases. These antibodies can promote immuno-stimulation of inherent immunological processes such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) or introduce a steric hindrance which blocks normal kinase functioning. Alternatively they can act as conjugate carriers for a variety of biologically active molecules, such as toxins, cytotoxic compounds, radioisotopes and enzymes. A collection of antibodies targeted to RTK extracellular domains, including cetuximab (Erbitux®), trastuzumab (Herceptin®), panitumumab (Vectibix®) and bevacizumab (Avastin®) have been developed and approved for the treatment of RTK-expressing cancers.
Trastuzumab is perhaps the most well-known and clinically significant RTK-targeted antibody; it is an engineered humanised IgG kappa light chain antibody, developed for use in the treatment of HER2 over-expressing breast cancers. The exact mechanism through which trastuzumab brings about its biological affects remains unclear however therapeutic benefits have been attributed to both F(ab)-mediated and Fc-facilitated functions.
Trastuzumab binding has been proposed to promote benefits including ADCC induction or HER2 ubiquitination, internalisation and degradation through activation of c-Cbl tyrosine kinase-ubiquitin ligase(22). Alternatively it has been suggested that trastuzumab promotes G1 cell cycle arrest through the up-regulation of cyclin-dependent kinase (CDK) inhibitor p27(1). Trastuzumab has also been credited with disrupting tyrosine-kinase mediated intracellular signalling by preventing HER2 receptor dimerization, consequently inhibiting Akt phosphorylation and blocking the PI3K/Akt signal transduction pathway. Furthermore, trastuzumab can block PI3Kinase signalling by increasing the membrane localisation and phosphatase activity of phosphatase and tensin homolog deleted on chromosome 10 (PTEN). PTEN prevents an accumulation of membrane-associated phosphatidylinositol triphosphate (PIP3) which acts as docking sites for downstream pleckstrin-homology (PH) domain containing proteins, such as Akt and PDK1(1,22).
Bevacizumab, a recombinant, humanised monoclonal antibody, specifically targets and binds to all isoforms of vascular endothelial growth factor-A (VEGF-A). Blocking VEGF-A, the preferred ligand of the RTKs, VEGFR 1 and 2 neutralises its potential to bind to and activate these receptors. An upregulation in the expression of both VEGF-A and VEGFR is linked to the induction of angiogenesis in highly vascularised tumour types. By binding and neutralising VEGF, bevacizumab blocks the potential of free VEGF to bind either receptor and confer downstream signalling, ultimately preventing the transcription of pro-angiogenic genes. Bevacizumab has been approved for the treatment of NSCLC, glioblastoma multiforme and metastatic colorectal cancer(23).
Novel and emerging targeting compounds
Despite the development of kinase inhibiting therapeutic drugs, the hunt for novel compounds which target kinase activity is still ongoing. Cancer’s ability to acquire drug resistances allows it to circumvent the suppressive effects of these RTK inhibiting drugs. Resistances, arising as mutations, can lead to constitutive receptor activation or up-regulations in alternative cell-signalling pathway components. These mutations ultimately render the suppressed kinases redundant and restore downstream-signalling potential. To this end modern pharmacology has turned to the deliberate search for new medicines from the natural world (termed biologicals or biologics). A variety of animal venoms, isolated from the classes Reptilia, Amphibia and the phylum Arthropoda, have been found to contain biologically active components that display antimicrobial, analgesic, anti-coagulant and anti-oncogenic potential. Although the use of venoms as therapeutics is still a fairly novel concept, emerging evidence already points to the potential for isolated components to be used in the targeted inhibition of RTK activity.
Cardiotoxin III is a basic 60aa polypeptide derived from the venom of Elapid Naja naja atra. It has been shown to display preferential cytotoxicity to cancerous cells through the inhibition of EGF/EGFR-mediated cell signalling pathways(24,25). Similar studies by Su et al. and Tsai et al. into cardiotoxin III’s potential to inhibit MDA-MB-231 breast cancer cells and A549 lung cancer cells respectively, showed that it effectively suppressed migration and invasiveness by blocking EGFR phosphorylation. Tsai et al. found cardiotoxin inhibited EGFR-phosphorylation, leading to ERK1/2 and PI3K/Akt downstream signal-pathways blockage, and ultimately decreased matrix metalloproteinase-9 expression(26). Alternatively Su et al showed EGFR-phosphorylation blockage led to reduced activation of downstream signal proteins PI3K/Akt, Janus tyrosine kinase-2 (JAK2) and activator of transcription STAT3(27).
Harnessing the power of nature
A pharmaceutical product currently under development, VRCTC-310-Onco, comprises both crotoxin (Crotalus durissus terrificus), a substance isolated from the South American rattlesnake, and cardiotoxin in equimolar ratios. Crotoxin, a non-covalent complex of two non-identical subunits, binds to and activates cellular surfaces receptors, particularly interfering with EGFR signalling(28). A study by Wang et al. into crotoxin showed interference with EGFR signalling. They observed stimulation of phosphorylation by crotoxin, however they were unable to establish the significance of this observation(29).
A study into the affects of alcoholic extracts from the centipede Scolopendra (AECS) by Ma et al. demonstrated its potential use in the inhibition of EGFR-dependent cancers. Experimental data showed that AECS inhibited cellular proliferation in high-EGFR expressing cell lines A431, HEK293 and HEK293/EGFR. Proposed mechanisms for this observation included the induction of apoptosis and the regulation of EGFR-dependent signalling cascades by altering EGFR-kinase phosphorylation and activity levels(30).
Meanwhile, a recent study by Li et al. investigated the potential of arenobufagin to inhibit angiogenic processes by targeting the VEGF/VEGFR-2 signalling pathways. Arenobufagin is a bufadienolide compound found to be secreted from the postauricular and skin glands of the toads Bufo gargarizans (Cantor) or Bufo melanostictus (Schneider). Experimental data showed that arenobufagin inhibited human umbilical vein endothelial cells’ (HUVECs) viability, cell migration and invasion. Similarly it prevented the angiogenic process of VEGF-induced vascular vessel sprout formation by inhibiting VEGFR-2 kinase activation. Arenobufagin inhibited VEGFR-2 phosphorylation and subsequently the downstream phosphorylation of Akt and mTOR without affecting their overall expression levels. Computational docking simulations suggested interactions between arenobufagin and the ATP-binding domain of VEGFR-2 kinase are responsible for these observations(31).
Despite evidence of compounds isolated from venoms for disease treatment, there is still little research taking place into venomous classes, particularly members of the arachnida. Within the arachnida, the family Theraphosidae (tarantulas) holds great prospects for the discovery of novel RTKs-targeted compounds, with approximately 900 theraphosid species discovered to date. To that end we have undertaken preliminary investigations into a panel of venoms isolated from theraphosids, using Alexa Fluor dye-labelled EGF competition assays. Initial investigations have identified 16 potential candidate venoms that displaced EGF binding and show potential as EGFR family inhibitors. In addition, further Western blot analysis into three of these candidate venoms demonstrated EGFR-phosphorylation inhibition in a dose-dependent manner. No effect on overall EGFR protein expression levels were observed, however these and many of the other venoms will ultimately warrant further investigations.
Bibliography
- Pohlmann PR, Mayer I a., Mernaugh R. Resistance to trastuzumab in breast cancer. Clin Cancer Res. 2009;15:7479–91
- Shin S-Y, Rath O, Choo S-M, Fee F, McFerran B, Kolch W, et al. Positive- and negative-feedback regulations coordinate the dynamic behavior of the Ras-Raf-MEK-ERK signal transduction pathway. J Cell Sci. 2009;122:425–35
- Lemmon M a., Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34
- Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther. 2011;11:777–92
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510
- Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4(1):127–50
- Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–84
- Wong K-K, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer therapy. Curr Opin Genet Dev. 2010;20(1):1–7
- Melnikova I, Golden J. Targeting protein kinases. Nat Rev Drug Discov. 2004;3(12):993–4
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol [Internet]. Nature Publishing Group; 2010;11(1):9–22. Available from: http://dx.doi.org/10.1038/nrm2822
- Hubbard SR, Miller TW. Receptor Tyrosine Kinases: Mechanisms of Activation and Signalling. Curr Opin Cell Biol. 2007;19(2):117–23
- Bazley L a., Gullick WJ. The epidermal growth factor receptor family. Endocr Relat Cancer. 2005;12:17–28
- Lee Y, Ma J, Lyu H, Huang J, Kim A, Liu B. Role of erbB3 receptors in cancer therapeutic resistance. Acta Biochim Biophys Sin (Shanghai). 2014;46(January):190–8
- Roskoski R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res [Internet]. Elsevier Ltd; 2014;79:34–74. Available from: http://dx.doi.org/10.1016/j.phrs.2013.11.002
- Roskoski R. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacol Res [Internet]. Elsevier Ltd; 2014;87:42–59. Available from: http://dx.doi.org/10.1016/j.phrs.2014.06.001
- Cataldo VD, Gibbons DL, Pérez-Soler R, Quintás-Cardama A. Treatment of non-small-cell lung cancer with erlotinib or gefitinib. N Engl J Med. 2011;364:947–55
- Rothschild SI, Gautschi O. Crizotinib in the treatment of non-small-cell lung cancer. Clin Lung Cancer [Internet]. Elsevier Inc; 2013;14(5):473–80. Available from: http://dx.doi.org/10.1016/j.cllc.2013.04.006
- Hallberg, Bengt P, Palmer, Ruth H P. Crizotinib — Latest Champion in the Cancer Wars ? N Engl J Med. 2010;1760–2
- Roy V, Perez E a. Beyond trastuzumab: small molecule tyrosine kinase inhibitors in HER-2-positive breast cancer. Oncologist. 2009;14:1061–9
- Wang Q, Quan H, Zhao J, Xie C, Wang L, Lou L. RON confers lapatinib resistance in HER2-positive breast cancer cells. Cancer Lett [Internet]. Elsevier Ireland Ltd; 2013;340(1):43–50. Available from: http://dx.doi.org/10.1016/j.canlet.2013.06.022
- Geuna E, Montemurro F, Aglietta M, Valabrega G. Potential of afatinib in the treatment of patients with HER2-positive breast cancer. Breast Cancer Targets Ther. 2012;4:131–7
- Vu T, Claret FX. Trastuzumab: Updated Mechanisms of Action and Resistance in Breast Cancer. Front Oncol. 2012;2(June):1–6
- Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis. 2012;15:171–85
- Su J-C, Lin K-L, Chien C-M, Chuang P-W, Chang L-S, Lin S-R. Concomitant inactivation of the epidermal growth factor receptor, phosphatidylinositol 3-kinase/Akt and Janus tyrosine kinase 2/signal transducer and activator of transcription 3 signalling pathways in cardiotoxin III-treated A549 cells. Clin Exp Pharmacol Physiol. 2010;37:833–40
- Gomes A, Bhattacharjee P, Mishra R, Biswas AK, Dasgupta SC, Giri B, et al. Anticancer potential of animal venoms and toxins. Indian J Exp Biol. 2010;48(February):93–103
- Tsai PC, Hsieh CY, Chiu CC, Wang CK, Chang L Sen, Lin SR. Cardiotoxin III suppresses MDA-MB-231 cell metastasis through the inhibition of EGF/EGFR-mediated signaling pathway. Toxicon. Elsevier Ltd; 2012;60(5):734–43
- Su JC, Lin KL, Chien CM, Chuang PW, Chang L Sen, Lin SR. Concomitant inactivation of the epidermal growth factor receptor, phosphatidylinositol 3-kinase/Akt and Janus tyrosine kinase 2/signal transducer and activator of transcription 3 signalling pathways in cardiotoxin III-treated A549 cells. Clin Exp Pharmacol Physiol. 2010;37:833–40
- Calderon L a, Sobrinho JC, Zaqueo KD, Moura A a De, Grabner AN, Mazzi M V, et al. Antitumoral Activity of Snake Venom Proteins : New Trends in Cancer Therapy. 2014;2014:1–19
- Wang JH, Xie Y, Wu JC, Han R, Reid PF, Qin ZH, et al. Crotoxin enhances the antitumor activity of gefinitib (Iressa) in SK-MES-1 human lung squamous carcinoma cells. Oncol Rep. 2012;27:1341–7
- Ma W, Zhang D, Zheng L, Zhan Y, Zhang Y. Potential roles of Centipede Scolopendra extracts as a strategy against EGFR-dependent cancers. 2015;7(1):39–52
- Li M, Wu S, Liu Z, Zhang W, Xu J, Wang Y, et al. Arenobufagin, a bufadienolide compound from toad venom, inhibits VEGF-mediated angiogenesis through suppression of VEGFR-2 signaling pathway. Biochem Pharmacol. Elsevier Inc.; 2012;83(9):1251–60
- Ou S-HI. Crizotinib: A novel and first-in-class multitargeted tyrosine kinase inhibitor for the treatment of anaplastic lymphoma kinase rearranged non-small cell lung cancer and beyond. Drug Des Devel Ther. 2011;5:471–85
Biographies
Danielle McCullough obtained a BSc (Honours) degree in biology and an MSc in Cancer Biology from the University of Kent, Canterbury. She has been a life science instructor at Canterbury Christ Church University since September 2014 and is currently undertaking a part-time PhD. The focus of her PhD is to investigate the receptor tyrosine kinase (RTK) inhibiting potential of venoms isolated from members of the theraphosids family of invertebrates, for the treatment of ErbB over-expressing cancer types.
Carol M. Trim received her PhD in Biochemistry from the University of Kent, Canterbury, investigating surrogate markers to determine drug activity of small molecule tyrosine kinase inhibitors. She then undertook eight years’ postdoctoral research at Kent first funded by a cancer charity and then collaborating with industry. She is now a senior lecturer in Biology at Canterbury Christ Church University investigating the use of venoms as potential as cancer therapies and anti-microbial drugs.